Wielowymiarowe role ciał ketonowych
Ciała ketonowe są tworzone przez wątrobę i wykorzystywane jako źródło energii, gdy glukoza nie jest łatwo dostępna w ludzkim ciele. Dwa główne ciała ketonowe to acetooctan (AcAc) i 3-beta-hydroksymaślan (3HB), podczas gdy aceton jest trzecim i najmniej występującym ciałem ketonowym. Ketony są zawsze obecne we krwi, a ich poziom wzrasta podczas postu i długotrwałych ćwiczeńketogenezy jest biochemicznym procesem, w którym organizmy produkują ciała ketonowe poprzez rozkład kwasów tłuszczowych i ketogennych aminokwasów.
Ciała ketonowe są generowane głównie w mitochondria komórek wątroby. Ketogeneza występuje, gdy poziom glukozy we krwi jest niski, szczególnie po wyczerpaniu innych komórkowych zapasów węglowodanów, takich jak glikogen. Mechanizm ten może również wystąpić, gdy nie ma wystarczającej ilości insuliny. Produkcja ciał ketonowych jest ostatecznie inicjowana w celu udostępnienia energii, która jest magazynowana w ludzkim ciele w postaci kwasów tłuszczowych. Ketogeneza zachodzi w mitochondriach, gdzie jest niezależnie regulowana.
Abstrakcyjny
Metabolizm ciała ketonowego jest centralnym węzłem homeostazy fizjologicznej. W tym przeglądzie omawiamy, w jaki sposób ketony służą dyskretnemu dostrajaniu ról metabolicznych, które optymalizują wydajność narządów i organizmu w zakresie różnych pozostałości składników odżywczych oraz chronią przed zapaleniem i urazami w wielu układach narządów. Tradycyjnie postrzegane jako substraty metaboliczne związane tylko z ograniczeniem węglowodanów, ostatnie obserwacje podkreślają znaczenie ciał ketonowych jako istotnych mediatorów metabolicznych i sygnalizacyjnych w przypadku obfitości węglowodanów. Uzupełniając repertuar znanych opcji terapeutycznych w chorobach układu nerwowego, pojawiły się potencjalne role ciał ketonowych w raku, a także intrygujące role ochronne w sercu i wątrobie, otwierając możliwości terapeutyczne w chorobach związanych z otyłością i układu krążenia. W celu pogodzenia klasycznego dogmatu ze współczesnymi obserwacjami omawiane są kontrowersje związane z metabolizmem i sygnalizacją ketonów.
Wprowadzenie
Ciała ketonowe są istotnym alternatywnym źródłem paliwa metabolicznego dla wszystkich dziedzin życia, eukarii, bakterii i archeonów (Aneja i in., 2002; Cahill GF Jr, 2006; Krishnakumar i in., 2008). Metabolizm ciał ketonowych u ludzi został wykorzystany do napędzania mózgu w okresowych okresach niedoboru składników odżywczych. Ciała ketonowe są przeplatane kluczowymi szlakami metabolicznymi ssaków, takimi jak a-utlenianie (FAO), cykl kwasów trikarboksylowych (TCA), glukoneogeneza, lipogeneza de novo (DNL) i biosynteza steroli. U ssaków ciała ketonowe są wytwarzane głównie w wątrobie z acetylo-CoA pochodzącego z FAO i są transportowane do tkanek pozawątrobowych w celu ostatecznego utlenienia. Ta fizjologia dostarcza alternatywnego paliwa, które jest wzmacniane przez stosunkowo krótkie okresy postu, co zwiększa dostępność kwasów tłuszczowych i zmniejsza dostępność węglowodanów (Cahill GF Jr, 2006; McGarry i Foster, 1980; Robinson i Williamson, 1980). Utlenianie ciała ketonowego staje się znaczącym czynnikiem przyczyniającym się do ogólnego metabolizmu energetycznego ssaków w tkankach pozawątrobowych w niezliczonych stanach fizjologicznych, w tym na czczo, głodzie, okresie noworodkowym, po wysiłku fizycznym, ciąży i przestrzeganiu diety niskowęglowodanowej. Całkowite stężenie ciał ketonowych w krążeniu u zdrowych dorosłych ludzi zwykle wykazuje wahania dobowe w zakresie około 100-250 μM, wzrasta do ~ 1 mM po długotrwałym wysiłku lub 24 godz. Na czczo i może kumulować się nawet do 20 mM w stanach patologicznych, takich jak cukrzycowa kwasica ketonowa ( Cahill GF Jr, 2006; Johnson i in., 1969b; Koeslag i in., 1980; Robinson i Williamson, 1980; Wildenhoff i in., 1974). Ludzka wątroba produkuje do 300 g ciał ketonowych dziennie (Balasse i Fery, 1989), które stanowią od 5% całkowitego wydatku energetycznego w stanach po posiłku, na czczo i z głodem (Balasse i in., 20; Cox i in. al., 1978).
Ostatnie badania podkreślają teraz rolę imperatywną dla ciał ketonowych w metabolizmie komórek ssaków, homeostazie i sygnalizacji w szerokim zakresie stanów fizjologicznych i patologicznych. Oprócz wykorzystywania jako paliwa energetyczne w tkankach pozawątrobowych, takich jak mózg, serce lub mięśnie szkieletowe, ciałka ketonowe odgrywają zasadniczą rolę jako mediatory sygnałowe, czynniki odpowiedzialne za potranslacyjną modyfikację białka (PTM) i modulatory stanu zapalnego i stres oksydacyjny. W tym przeglądzie przedstawiamy klasyczne i współczesne poglądy o plejotropowej roli ciał ketonowych i ich metabolizmu.
Przegląd metabolizmu ciała ketonowego
Tempo ketogenezy wątrobowej jest regulowane przez serię fizjologicznych i biochemicznych przemian tłuszczu. Podstawowe regulatory obejmują lipolizę kwasów tłuszczowych z triacylogliceroli, transport do i przez błonę plazmatyczną hepatocytów, transport do mitochondriów za pośrednictwem palmitoilotransferazy karnityny 1 (CPT1), spiralę P-oksydacji, aktywność cyklu TCA i stężenia pośrednie, potencjał redoks i regulatory hormonalne tych procesów, głównie glukagonu i insuliny [przegląd w (Arias i wsp., 1995; Ayte i wsp., 1993; Ehara i wsp., 2015; Ferre i wsp., 1983; Kahn i wsp., 2005; McGarry i Foster , 1980; Williamson i in., 1969)]. Klasycznie ketogeneza jest postrzegana jako szlak pośredni, w którym acetylo-CoA pochodzący z a-utleniania przewyższa aktywność syntazy cytrynianowej i/lub dostępność szczawiooctanu do kondensacji z wytworzeniem cytrynianu. Trójwęglowe produkty pośrednie wykazują aktywność antyketogenną, prawdopodobnie ze względu na ich zdolność do powiększania puli szczawiooctanów w celu spożycia acetylo-CoA, ale samo stężenie acetylo-CoA w wątrobie nie determinuje szybkości ketogenicznej (Foster, 1967; Rawat i Menahan, 1975; Williamson i in., 1969). Regulacja ketogenezy przez zdarzenia hormonalne, transkrypcyjne i potranslacyjne razem wspierają pogląd, że mechanizmy molekularne, które dostrajają tempo ketogenu, pozostają nie w pełni poznane (patrz Regulacja HMGCS2 i SCOT/OXCT1).
Ketogeneza zachodzi głównie w macierzy mitochondrialnej wątroby z szybkością proporcjonalną do całkowitego utleniania tłuszczu. Po transporcie łańcuchów acylowych przez błony mitochondrialne i p-oksydacji, izoforma mitochondrialna syntazy 3-hydroksymetyloglutarylo-CoA (HMGCS2) katalizuje los powodując kondensację acetoacetylo-CoA (AcAc-CoA) i acetylo-CoA w celu wytworzenia HMG-CoA (Rys. 1A). Liaza HMG-CoA (HMGCL) rozszczepia HMG-CoA, aby uwolnić acetylo-CoA i acetooctan (AcAc), a ten ostatni jest redukowany do d - a - hydroksymaślanu (d-? OHB) przez zależną od fosfatydylocholiny mitochondrialną dehydrogenazę d-aOHB ( BDH1) w reakcji bliskiej równowagi sprzężonej z NAD + / NADH (Bock i Fleischer, 1975; LEHNINGER i wsp., 1960). Stała równowagi BDH1 sprzyja wytwarzaniu d-a-OHB, ale stosunek ciał ketonowych AcAc / d-a-OHB jest wprost proporcjonalny do mitochondrialnego stosunku NAD + / NADH, a zatem aktywność oksydoreduktazy BDH1 moduluje mitochondrialny potencjał redoks (Krebs i wsp., 1969; Williamson i wsp., 1967). AcAc może również spontanicznie dekarboksylować do acetonu (Pedersen, 1929), źródła słodkiego zapachu u ludzi cierpiących na kwasicę ketonową (tj. Całkowita zawartość ciał ketonowych w surowicy> ~ 7 mM; AcAc pKa 3.6, OHB pKa 4.7). Mechanizmy transportu ciał ketonowych przez wewnętrzną błonę mitochondrialną nie są znane, ale AcAc / d-aOHB są uwalniane z komórek za pośrednictwem transporterów monokarboksylanów (u ssaków MCT 1 i 2, znane również jako członkowie rodziny substancji rozpuszczonych 16A 1 i 7) i transportowany w krążeniu do tkanek pozawątrobowych w celu ostatecznego utleniania (Cotter i wsp., 2011; Halestrap i Wilson, 2012; Halestrap, 2012; Hugo i wsp., 2012). Stężenia krążących ciał ketonowych są wyższe niż w tkankach pozawątrobowych (Harrison i Long, 1940), co wskazuje, że ciała ketonowe są transportowane w dół gradientu stężeń. Mutacje powodujące utratę funkcji w MCT1 są związane ze spontanicznymi napadami kwasicy ketonowej, co sugeruje krytyczną rolę w imporcie ciał ketonowych.

� Z wyjątkiem potencjalnego przekierowania ciał ketonowych na nieoksydacyjne losy (patrz Nieoksydacyjne metaboliczne losy ciał ketonowych), hepatocyty nie mają zdolności do metabolizowania wytwarzanych przez nie ciał ketonowych. Ciała ketonowe syntetyzowane de novo przez wątrobę są (i) katabolizowane w mitochondriach tkanek pozawątrobowych do acetylo-CoA, który jest dostępny w cyklu TCA do końcowego utleniania (ryc. 1A), (ii) przekierowany do szlaków lipogenezy lub syntezy steroli ( Ryc. 1B) lub (iii) wydalane z moczem. Jako alternatywne paliwo energetyczne ciała ketonowe są intensywnie utleniane w sercu, mięśniach szkieletowych i mózgu (Balasse i Fery, 1989; Bentourkia i in., 2009; Owen i in., 1967; Reichard i in., 1974; Sultan, 1988 ). Pozawątrobowa mitochondrialna BDH1 katalizuje pierwszą reakcję utleniania -OHB, przekształcając ją w AcAc (LEHNINGER i wsp., 1960; Sandermann i wsp., 1986). Cytoplazmatyczna dehydrogenaza d-AOHB (BDH2) z tylko 20% identycznością sekwencji z BDH1 ma wysoką Km dla ciał ketonowych, a także odgrywa rolę w homeostazie żelaza (Davuluri i wsp., 2016; Guo i wsp., 2006) . W pozawątrobowej macierzy mitochondrialnej AcAc jest aktywowany do AcAc-CoA poprzez wymianę ugrupowania CoA z sukcynylo-CoA w reakcji katalizowanej przez unikalną ssaczą transferazę CoA, transferazę sukcynylo-CoA:3-oksokwas-CoA (SCOT, transferaza CoA; kodowane przez OXCT1), w wyniku reakcji bliskiej równowadze. Energia swobodna uwalniana przez hydrolizę AcAc-CoA jest większa niż energia sukcynylo-CoA, co sprzyja tworzeniu AcAc. Tak więc przepływ oksydacyjny ciała ketonowego następuje w wyniku działania masowego: obfita podaż AcAc i szybkie zużycie acetylo-CoA przez syntazę cytrynianową sprzyja tworzeniu AcAc-CoA (+ bursztynian) przez SCOT. Warto zauważyć, że w przeciwieństwie do glukozy (heksokinazy) i kwasów tłuszczowych (syntetazy acylo-CoA), aktywacja ciał ketonowych (SCOT) do postaci ulegającej utlenieniu nie wymaga inwestycji ATP. Odwracalna reakcja tiolazy AcAc-CoA [katalizowana przez dowolną z czterech mitochondrialnych tiolaz kodowanych przez ACAA2 (kodujący enzym znany jako T1 lub CT), ACAT1 (kodujący T2), HADHA lub HADHB] daje dwie cząsteczki acetylo-CoA, które wchodzą w cykl TCA (Hersh i Jencks, 1967; Stern i in., 1956; Williamson i in., 1971). Podczas stanów ketonowych (tj. całkowita ilość ketonów w surowicy > 500 μM), ciała ketonowe stają się znaczącymi uczestnikami wydatkowania energii i są szybko wykorzystywane w tkankach, aż do wychwytu lub nasycenia utleniania (Balasse i wsp., 1978; Balasse i Fery, 1989 Edmond i wsp., 1987). Bardzo małą część ciał ketonowych pochodzących z wątroby można łatwo zmierzyć w moczu, a wskaźniki wykorzystania i reabsorpcji przez nerki są proporcjonalne do stężenia w krążeniu (Goldstein, 1987; Robinson i Williamson, 1980). Podczas stanów wysoce ketotycznych (> 1 mM w osoczu), ketonuria służy jako półilościowy reporter ketozy, chociaż większość testów klinicznych ciał ketonowych w moczu wykrywa AcAc, ale nie ?OHB (Klocker i wsp., 2013).
Substraty ketogeniczne i ich wpływ na metabolizm hepatocytów
Substancje ketogenne obejmują kwasy tłuszczowe i aminokwasy (ryc. 1B). Katabolizm aminokwasów, w szczególności leucyny, generuje około 4% ciał ketonowych w stanie poabsorpcyjnym (Thomas i wsp., 1982). Tak więc pula substratu acetylo-CoA do generowania ciał ketonowych pochodzi głównie z kwasów tłuszczowych, ponieważ w stanach zmniejszonego dostarczania węglowodanów pirogronian wchodzi do cyklu wątrobowego TCA głównie poprzez anaplerozę, tj. Zależną od ATP karboksylację do szczawiooctanu (OAA) lub do jabłczanu (MAL), a nie oksydacyjnej dekarboksylacji do acetylo-CoA (Jeoung i wsp., 2012, Magnusson i wsp., 1991, Merritt i wsp., 2011). W wątrobie glukoza i pirogronian przyczyniają się w sposób nieistotny do ketogenezy, nawet gdy maksymalna dekarboksylacja pirogronianu do acetylo-CoA jest największa (Jeoung i wsp., 2012).
Acetyl-CoA obejmuje kilka ról stanowiących integralną część wątrobowego metabolizmu pośredniego poza generowaniem ATP poprzez końcowe utlenianie (patrz także: Integracja metabolizmu ciała ketonowego, modyfikacja potranslacyjna i fizjologia komórki). Acetyl-CoA allosterycznie aktywuje (i) karboksylazę pirogronianową (PC), aktywując tym samym mechanizm metabolicznej kontroli, który zwiększa anaplerotyczne wejście metabolitów do cyklu TCA (Owen i wsp., 2002, Scrutton i Utter, 1967) i (ii) dehydrogenazę pirogronianową kinaza, która fosforyluje i hamuje dehydrogenazę pirogronianową (PDH) (Cooper i wsp., 1975), co dodatkowo zwiększa przepływ pirogronianu do cyklu TCA poprzez anapterozę. Ponadto, cytoplazmatyczny acetylo-CoA, którego pula jest wzmacniana przez mechanizmy, które przekształcają mitochondrialny acetylo-CoA w transportowalne metabolity, hamuje utlenianie kwasów tłuszczowych: karboksylaza acetyl-CoA (ACC) katalizuje konwersję acetylo-CoA do malonylo-CoA, lipogennego substratu i allosteryczny inhibitor mitochondrialnego CPT1 [omówionego w (Kahn i wsp., 2005, McGarry and Foster, 1980)]. W ten sposób mitochondrialna pula acetylo-CoA reguluje i jest regulowana przez szlak spillover ketogenezy, który koordynuje kluczowe aspekty wątrobowego pośredniego metabolizmu.
Nieoksydacyjne metaboliczne losy ciał ketonowych
Dominującym losem ketonów pochodzących z wątroby jest pozawątrobowe utlenianie zależne od SCOT. Jednak AcAc może być eksportowany z mitochondriów i wykorzystywany w szlakach anabolicznych poprzez konwersję do AcAc-CoA w reakcji zależnej od ATP katalizowanej przez cytoplazmatyczną syntetazę acetoacetylo-CoA (AACS, Fig. 1B). Szlak ten jest aktywny podczas rozwoju mózgu oraz w okresie laktacji (Morris, 2005; Robinson i Williamson, 1978; Ohgami i in., 2003). AACS jest również silnie wyrażany w tkance tłuszczowej i aktywowanych osteoklastach (Aguilo i wsp., 2010; Yamasaki i wsp., 2016). Cytoplazmatyczny AcAc-CoA może być kierowany przez cytozolowy HMGCS1 w kierunku biosyntezy steroli lub rozszczepiany przez jedną z dwóch tiolaz cytoplazmatycznych do acetylo-CoA (ACAA1 i ACAT2), karboksylowany do malonylo-CoA i przyczyniać się do syntezy kwasów tłuszczowych (Bergstrom et al. wsp., 1984; Edmond, 1974; Endemann i wsp., 1982; Geelen i wsp., 1983; Webber i Edmond, 1977).
Chociaż znaczenie fizjologiczne nie zostało jeszcze ustalone, ketony mogą służyć jako substraty anaboliczne nawet w wątrobie. W sztucznych kontekstach eksperymentalnych AcAc może przyczyniać się do powstania nawet połowy nowo zsyntetyzowanego lipidu i do 75% nowego zsyntetyzowanego cholesterolu (Endemann i wsp., 1982; Geelen i wsp., 1983; Freed i wsp., 1988). Ponieważ AcAc pochodzi z niecałkowitego utleniania tłuszczu w wątrobie, zdolność AcAc do udziału w lipogenezie in vivo sugerowałaby jałowy cykl wątrobowy, w którym ketony pochodzące z tłuszczów mogą być wykorzystywane do produkcji lipidów, co jest pojęciem, którego znaczenie fizjologiczne wymaga walidacji eksperymentalnej, ale może służyć role adaptacyjne lub nieadaptacyjne (Solinas i in., 2015). AcAc chciwie dostarcza cholesterogenezę, z niskim AACS Km-AcAc (~50 ?M) sprzyjającym aktywacji AcAc nawet po posiłku (Bergstrom i wsp., 1984). Zasugerowano dynamiczną rolę cytoplazmatycznego metabolizmu ketonów w pierwotnych neuronach embrionalnych myszy oraz w adipocytach pochodzących z 3T3-L1, ponieważ knockdown AACS zaburza różnicowanie każdego typu komórek (Hasegawa i wsp., 2012a; Hasegawa i wsp., 2012b). Powalenie AACS u myszy in vivo obniżyło poziom cholesterolu w surowicy (Hasegawa i wsp., 2012c). SREBP-2, główny regulator transkrypcji biosyntezy cholesterolu i receptor aktywowany przez proliferatory peroksysomów (PPAR)-? są aktywatorami transkrypcji AACS i regulują jego transkrypcję podczas rozwoju neurytów oraz w wątrobie (Aguilo i wsp., 2010; Hasegawa i wsp., 2012c). Podsumowując, cytoplazmatyczny metabolizm ciał ketonowych może być ważny w wybranych stanach lub naturalnej historii choroby, ale nie jest wystarczający do usuwania ciał ketonowych pochodzących z wątroby, ponieważ masywna hiperketonemia występuje w warunkach selektywnego upośledzenia pierwotnego losu oksydacyjnego poprzez mutacje utraty funkcji SCOT (Berry i in., 2001; Cotter i in., 2011).
Regulacja HMGCS2 i SCOT / OXCT1
Odejście mitochondrialnego od genu kodującego cytozolowy HMGCS nastąpiło na wczesnym etapie ewolucji kręgowców ze względu na potrzebę wspierania ketogenezy wątrobowej u gatunków o wyższym stosunku masy mózgu do masy ciała (Boukaftane i wsp., 1994; Cunnane i Crawford, 2003). Naturalnie występujące mutacje utraty funkcji HMGCS2 u ludzi powodują napady hipoglikemii hipoketotycznej (Pitt i wsp., 2015; Thompson i wsp., 1997). Silna ekspresja HMGCS2 jest ograniczona do hepatocytów i nabłonka okrężnicy, a jego ekspresja i aktywność enzymatyczna są koordynowane przez różne mechanizmy (Mascaro i wsp., 1995; McGarry i Foster, 1980; Robinson i Williamson, 1980). Podczas gdy pełny zakres stanów fizjologicznych wpływających na HMGCS2 wymaga dalszego wyjaśnienia, jego ekspresja i/lub aktywność jest regulowana we wczesnym okresie poporodowym, starzenia się, cukrzycy, głodzie lub przyjmowaniu diety ketogenicznej (Balasse i Fery, 1989; Cahill GF Jr, 2006 Girard i in., 1992; Hegardt, 1999; Satapati i in., 2012; Sengupta i in., 2010). U płodu metylacja regionu flankującego 5� genu Hmgcs2 jest odwrotnie skorelowana z jego transkrypcją i ulega częściowemu odwróceniu po urodzeniu (Arias i wsp., 1995; Ayte i wsp., 1993; Ehara i wsp., 2015; Ferre i wsp., 1983; ., XNUMX). Podobnie, wątrobowe Bdh1 wykazuje rozwojowy wzorzec ekspresji, wzrastający od urodzenia do odsadzenia, a także jest indukowany przez dietę ketogeniczną w sposób zależny od czynnika wzrostu fibroblastów (FGF)-21 (Badman i wsp., 2007; Zhang i wsp., 1989 ). Ketogeneza u ssaków jest wysoce wrażliwa zarówno na insulinę, jak i glukagon, które są odpowiednio tłumione i stymulowane (McGarry i Foster, 1977). Insulina hamuje lipolizę tkanki tłuszczowej, tym samym pozbawiając ketogenezę jej substratu, podczas gdy glukagon zwiększa przepływ ketogenny poprzez bezpośredni wpływ na wątrobę (Hegardt, 1999). Transkrypcja Hmgcs2 jest stymulowana przez przedni czynnik transkrypcyjny FOXA2, który jest hamowany przez kinazę insulinowo-fosfatydyloinozytol-3/Akt i jest indukowany przez sygnalizację glukagon-cAMP-p300 (Arias i in., 1995; Hegardt, 1999; Quant i in. , 1990; Thumelin i wsp. 1993; von Meyenn i wsp. 2013; Wolfrum i wsp. 2004; Wolfrum i wsp. 2003). PPAR? (Rodriguez i wsp., 1994) wraz ze swoim celem, FGF21 (Badman i wsp., 2007) również indukują transkrypcję Hmgcs2 w wątrobie podczas głodzenia lub podawania diety ketogenicznej (Badman i wsp., 2007; Inagaki i wsp., 2007 ). Indukcja PPAR? może wystąpić przed przejściem z fizjologii płodowej do noworodkowej, podczas gdy aktywacja FGF21 może być faworyzowana we wczesnym okresie noworodkowym poprzez hamowanie deacetylazy histonowej (HDAC)-3, w której pośredniczy ?OHB (Rando i wsp., 2016). zależne od mTORC1 (ssaczego celu kompleksu rapamycyny 1) hamowanie PPAR? aktywność transkrypcyjna jest również kluczowym regulatorem ekspresji genu Hmgcs2 (Sengupta i wsp., 2010), a wątrobowy PER2, główny oscylator dobowy, pośrednio reguluje ekspresję Hmgcs2 (Chavan i wsp., 2016). Ostatnie obserwacje wskazują, że interleukina-6 wywołana przez nowotwór pozawątrobowy zaburza ketogenezę poprzez PPAR? tłumienie (Flint i in., 2016).
Aktywność enzymu HMGCS2 reguluje się za pomocą wielu PTM. Fosforylacja seryny HMGCS2 wzmacniała jej aktywność in vitro (Grimsrud i wsp., 2012). Aktywność HMGCS2 jest allosterycznie hamowana sukcynylacją sukcynylo-CoA i lizyny (Arias i wsp., 1995, Hegardt, 1999, Lowe i Tubbs, 1985, Quant i wsp., 1990, Rardin i wsp., 2013, Reed i wsp., 1975; Thumelin i wsp., 1993). Sukcynylacja reszt lizyny HMGCS2, HMGCL i BDH1 w mitochondriach wątrobowych jest celami zależnej od NAD + deacylazy sirtuiny 5 (SIRT5) (Rardin i wsp., 2013). Aktywność HMGCS2 jest również zwiększona przez deacetylację lizyny SIRT3 i możliwe jest, że przesłuch między acetylacją a sukcynylacją reguluje aktywność HMGCS2 (Rardin i wsp., 2013, Shimazu i wsp., 2013). Pomimo zdolności tych PTM do regulowania HMGCS2 Km i Vmax, fluktuacje tych PTM nie zostały jeszcze dokładnie odwzorowane i nie zostały potwierdzone jako mechanistyczne czynniki ketogenezy in vivo.
SCOT ulega ekspresji we wszystkich komórkach ssaków, które zawierają mitochondria, z wyjątkiem komórek hepatocytów. Znaczenie aktywności SCOT i ketolizy wykazano u myszy SCOT-KO, które wykazywały jednolitą śmiertelność z powodu hipoglikemii hiperketonemicznej w ciągu 48 godzin po urodzeniu (Cotter i wsp., 2011). Specyficzna tkankowa utrata SCOT w neuronach lub miocytach szkieletowych wywołuje nieprawidłowości metaboliczne podczas głodu, ale nie jest śmiertelna (Cotter i wsp., 2013b). U ludzi niedobór SCOT objawia się we wczesnym okresie życia ciężką kwasicą ketonową, powodując letarg, wymioty i śpiączkę (Berry i in., 2001; Fukao i in., 2000; Kassovska-Bratinova i in., 1996; Niezen-Koning i in. , 1997; Saudubray i wsp., 1987; Snyderman i wsp., 1998; Tildon i Cornblath, 1972). Stosunkowo niewiele wiadomo na poziomie komórkowym na temat regulatorów ekspresji genu SCOT i białek. Ekspresja mRNA Oxct1 oraz białko i aktywność SCOT są zmniejszone w stanach ketotycznych, prawdopodobnie przez mechanizmy zależne od PPAR (Fenselau i Wallis, 1974; Fenselau i Wallis, 1976; Grinblat i wsp., 1986; Okuda i wsp., 1991; Turko i wsp., 2001; Grinblat i wsp., 2010; ., 1; Wentz i in., 2). W cukrzycowej kwasicy ketonowej niedopasowanie między wątrobową ketogenezą a pozawątrobowym utlenianiem nasila się z powodu upośledzenia aktywności SCOT. Nadekspresja niezależnego od insuliny transportera glukozy (GLUT1/SLC1A2009) w kardiomiocytach również hamuje ekspresję genu Oxct1 i obniża końcowe utlenianie ketonów w stanie nieketotycznym (Yan i wsp., 122). W wątrobie obfitość mRNA Oxct3 jest tłumiona przez mikroRNA-27 i metylację histonów H3K2011me1, które są widoczne podczas przejścia z okresu płodowego do okresu noworodkowego (Thorrez i wsp., 1). Jednak tłumienie ekspresji Oxct1 w wątrobie w okresie poporodowym można przede wszystkim przypisać usunięciu krwiotwórczych komórek progenitorowych z ekspresją Oxct1 z wątroby, a nie utracie wcześniej istniejącej ekspresji Oxct2008 w terminalnie zróżnicowanych hepatocytach. W rzeczywistości ekspresja mRNA OxctXNUMX i białka SCOT w zróżnicowanych hepatocytach jest niezwykle niska (Orii i wsp., XNUMX).
SCOT jest również regulowany przez PTM. Enzym jest hiperacetylowany w mózgach myszy SIRT3 KO, które również wykazują zmniejszoną produkcję acetylo-CoA zależną od AcAc (Dittenhafer-Reed et al., 2015). Nieenzymatyczne nitrowanie reszt tyrozynowych SCOT również osłabia jego aktywność, o czym donoszono w sercach różnych modeli myszy z cukrzycą (Marcondes i wsp., 2001; Turko i wsp., 2001; Wang i wsp., 2010a). W przeciwieństwie do tego azotowanie reszty tryptofanu zwiększa aktywność SCOT (Br g re i in., 2010; Rebrin i in., 2007). Mogą istnieć molekularne mechanizmy nitrowania lub de-nitracji specyficzne dla pozostałości, mające na celu modulowanie aktywności SCOT i wymagają wyjaśnienia.
Kontrowersje w ketogenezie pozawotworowej
U ssaków głównym organem ketogennym jest wątroba i tylko hepatocyty i komórki nabłonka jelita w znacznym stopniu wyrażają mitochondrialną izoformę HMGCS2 (Cotter i wsp., 2013a; Cotter i wsp., 2014; McGarry i Foster, 1980; Robinson i Williamson, 1980) . Beztlenowa fermentacja bakteryjna złożonych polisacharydów daje maślan, który jest absorbowany przez kolonocyty u ssaków w celu ostatecznego utlenienia lub ketogenezy (Cherbuy i wsp., 1995), co może odgrywać rolę w różnicowaniu kolonocytów (Wang i wsp., 2016). Z wyłączeniem komórek nabłonka jelita i hepatocytów, HMGCS2 jest prawie nieobecny w prawie wszystkich innych komórkach ssaków, ale perspektywa pozawątrobowej ketogenezy została zwiększona w komórkach nowotworowych, astrocytach ośrodkowego układu nerwowego, nerkach, trzustce? komórki, nabłonek barwnikowy siatkówki (RPE), a nawet w mięśniach szkieletowych (Adijanto i wsp., 2014; Avogaro i wsp., 1992; El Azzouny i wsp., 2016; Grabacka i wsp., 2016; Kang i wsp., 2015 ; Le Foll i in., 2014; Nonaka i in., 2016; Takagi i in., 2016a; Thevenet i in., 2016; Zhang i in., 2011). Ektopowy HMGCS2 obserwowano w tkankach, które nie mają netto zdolności ketogennej (Cook i in., 2016; Wentz i in., 2010), a HMGCS2 wykazuje prospektywną aktywność `` oświetlenia księżyca '' niezależną od ketogenezy, w tym w jądrze komórkowym (Chen i wsp. , 2016; Kostiuk i in., 2010; Meertens i in., 1998).
Każda tkanka pozawątrobowa, która utlenia ciała ketonowe, ma również potencjał do gromadzenia ciał ketonowych poprzez niezależne mechanizmy HMGCS2 (ryc. 2A). Jednak nie ma tkanki pozawątrobowej, w której stężenie ciał ketonowych w stanie ustalonym przewyższa stężenie w krążeniu (Cotter i in., 2011; Cotter i in., 2013b; Harrison i Long, 1940), podkreślając, że ciała ketonowe są transportowane w dół gradient stężenia poprzez mechanizmy zależne od MCT1 / 2. Jeden mechanizm pozawątrobowej ketogenezy może w rzeczywistości odzwierciedlać względne upośledzenie utleniania ketonów. Dodatkowe potencjalne wyjaśnienia dotyczą dziedziny tworzenia się ciał ketonowych. Po pierwsze, ketogeneza de novo może zachodzić poprzez odwracalną aktywność enzymatyczną tiolazy i SCOT (Weidemann i Krebs, 1969). Gdy stężenie acetylo-CoA jest stosunkowo wysokie, reakcje normalnie odpowiedzialne za utlenianie AcAc działają w odwrotnym kierunku (GOLDMAN, 1954). Drugi mechanizm występuje, gdy związki pośrednie pochodzące z β-utleniania gromadzą się z powodu wąskiego gardła cyklu TCA, AcAc-CoA przekształca się w l-a-OHB-CoA w wyniku reakcji katalizowanej przez mitochondrialną dehydrogenazę 3-hydroksyacylo-CoA, a następnie przez 3-hydroksybutyryl Deacylazę CoA do l-aOHB, której nie można odróżnić metodą spektrometrii mas lub spektroskopii rezonansowej od fizjologicznego enancjomeru d-aOHB (Reed i Ozand, 1980). I-aOHB można odróżnić chromatograficznie lub enzymatycznie od d-aOHB i występuje w tkankach pozawątrobowych, ale nie w wątrobie ani we krwi (Hsu i wsp., 2011). Ketogeneza wątrobowa wytwarza tylko d-aOHB, jedyny enancjomer będący substratem BDH (Ito i wsp., 1984; Lincoln i wsp., 1987; Reed i Ozand, 1980; Scofield i wsp., 1982; Scofield i wsp., 1982). Trzeci mechanizm niezależny od HMGCS2 generuje d-aOHB poprzez katabolizm aminokwasów, szczególnie leucyny i lizyny. Czwarty mechanizm jest pozorny tylko dlatego, że jest spowodowany artefaktem znakowania i dlatego jest nazywany pseudoketogenezą. Zjawisko to można przypisać odwracalności reakcji SCOT i tiolazy i może powodować przeszacowanie obrotu ciała ketonowego z powodu izotopowego rozcieńczenia ciała ketonowego znacznika w tkance pozawątrobowej (Des Rosiers i wsp., 1990; Fink i wsp., 1988) . Niemniej jednak pseudoketogeneza może być pomijalna w większości kontekstów (Bailey i in., 1990; Keller i in., 1978). Schemat (ryc. 2A) wskazuje przydatne podejście do zastosowania, biorąc pod uwagę podwyższone stężenie ketonów w tkankach w stanie stacjonarnym.


� Ostatnio zwrócono uwagę na nerkę jako potencjalnie ketogenny narząd. W ogromnej większości stanów nerka jest konsumentem netto ciał ketonowych pochodzenia wątrobowego, wydalających lub ponownie wchłaniających ciała ketonowe z krwioobiegu, a nerki na ogół nie są generatorem ani koncentratorem ciał ketonowych netto (Robinson i Williamson, 1980). Autorzy klasycznego badania doszli do wniosku, że minimalna ketogeneza nerkowa określona ilościowo w sztucznym układzie doświadczalnym nie była fizjologicznie istotna (Weidemann i Krebs, 1969). Ostatnio w mysich modelach z cukrzycą i niedoborem autofagii wywnioskowano ketogenezę nerkową, ale bardziej prawdopodobne jest, że wielonarządowe przesunięcia w homeostazie metabolicznej zmieniają integracyjny metabolizm ketonów poprzez oddziaływanie na wiele narządów (Takagi i wsp., 2016a; Takagi i wsp., 2016b; Zhang i in., 2011). Jedna z niedawnych publikacji sugerowała ketogenezę nerkową jako mechanizm ochronny przed uszkodzeniem niedokrwienno-reperfuzyjnym w nerkach (Tran i wsp., 2016). Bezwzględne stężenia αOHB w stanie stacjonarnym z ekstraktów tkanki nerki myszy odnotowano przy ~4 mM. Aby sprawdzić, czy jest to wykonalne, obliczyliśmy ilościowo stężenia αOHB w ekstraktach nerkowych myszy karmionych i głodzonych 12 godziny na dobę. Stężenie ?OHB w surowicy wzrosło z ~24 μM do 100 mM po 2 godzinach na czczo (ryc. 24B), podczas gdy stężenia αOHB w stanie stacjonarnym w nerkach około 2 μM po posiłku i tylko 100 mM po 1 godzinach na czczo (ryc. 24C�E), obserwacje zgodne ze stężeniami obliczonymi ponad 2 lat temu (Hems i Brosnan, 45). Pozostaje możliwe, że w stanach ketonowych ciała ketonowe pochodzenia wątrobowego mogą działać renoprotekcyjnie, ale dowody na ketogenezę nerek wymagają dalszego uzasadnienia. Przekonujące dowody potwierdzające prawdziwą ketogenezę pozawątrobową przedstawiono w RPE (Adijanto i wsp., 1970). Zasugerowano, że ta intrygująca przemiana metaboliczna potencjalnie umożliwi ketonom pochodzącym z RPE przepływ do fotoreceptorów lub komórek gleju Mellera, co może pomóc w regeneracji zewnętrznego segmentu fotoreceptora.
„OHB jako mediator sygnalizujący”
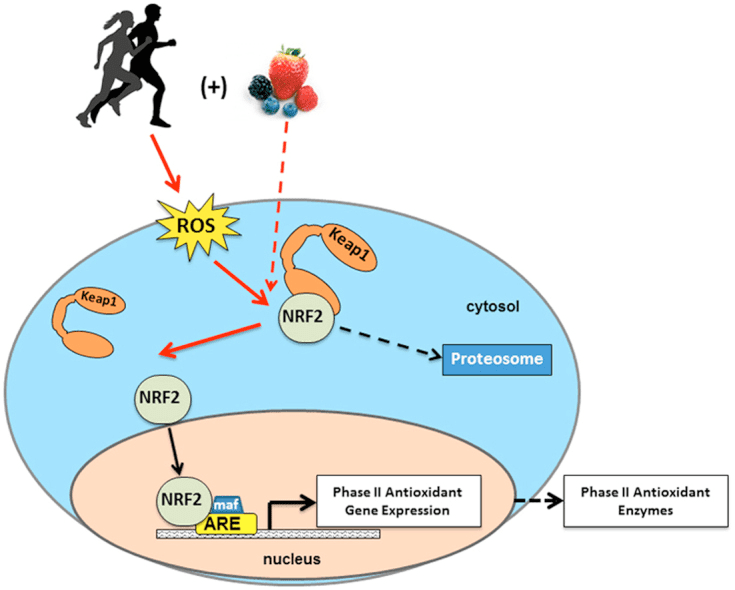
Chociaż są one bogate energetycznie, ciała ketonowe pełnią prowokacyjne "niekanoniczne" role sygnalizacyjne w homeostazie komórkowej (ryc. 3) (Newman i Verdin, 2014; Rojas-Morales i in., 2016). Na przykład, aOHB hamuje HDAC klasy I, co zwiększa acetylację histonów i tym samym indukuje ekspresję genów, które ograniczają stres oksydacyjny (Shimazu i wsp., 2013). ?OHB sam w sobie jest kowalencyjnym modyfikatorem histonów w resztach lizyny w wątrobach myszy z cukrzycą na czczo lub myszy z cukrzycą indukowaną streptozotocyną (Xie i wsp., 2016) (patrz również poniżej, Integracja metabolizmu ciał ketonowych, modyfikacja potranslacyjna i fizjologia komórki, oraz Ciała ketonowe, stres oksydacyjny i neuroprotekcja).

?OHB jest również efektorem poprzez receptory sprzężone z białkiem G. Poprzez niejasne mechanizmy molekularne hamuje aktywność współczulnego układu nerwowego i zmniejsza całkowite zużycie energii i tętno poprzez hamowanie sygnalizacji krótkołańcuchowych kwasów tłuszczowych przez receptor 41 sprzężony z białkiem G (GPR41) (Kimura i wsp., 2011). Jeden z najlepiej zbadanych efektów sygnalizacyjnych aOHB zachodzi przez GPR109A (znany również jako HCAR2), członka podrodziny kwasu hydrokarboksylowego GPCR ulegającego ekspresji w tkance tłuszczowej (białej i brązowej) (Tunaru i wsp., 2003) oraz w komórki odpornościowe (Ahmed i wsp., 2009). AOHB jest jedynym znanym endogennym ligandem receptora GPR109A (EC50 ~770 µM) aktywowanym przez d-AOHB, l-AOHB i maślan, ale nie AcAc (Taggart i wsp., 2005). Wysoki próg stężenia dla aktywacji GPR109A osiąga się poprzez przestrzeganie diety ketogenicznej, głód lub podczas kwasicy ketonowej, co prowadzi do zahamowania lipolizy tkanki tłuszczowej. Działanie antylipolityczne GPR109A przebiega poprzez hamowanie cyklazy adenylylowej i zmniejszenie cAMP, hamując wrażliwą na hormony lipazę triglicerydową (Ahmed i wsp., 2009; Tunaru i wsp., 2003). Tworzy to pętlę ujemnego sprzężenia zwrotnego, w której ketoza hamuje modulację ketogenezy poprzez zmniejszenie uwalniania niezestryfikowanych kwasów tłuszczowych z adipocytów (Ahmed et al., 2009; Taggart et al., 2005), efekt, który można zrównoważyć przez napęd współczulny, który stymuluje lipolizę. Niacyna (witamina B3, kwas nikotynowy) jest silnym (EC50 ~ 0.1 M) ligandem dla GRP109A, skutecznie stosowanym od dziesięcioleci w dyslipidemii (Benyo i wsp., 2005; Benyo i wsp., 2006; Fabbrini i wsp., 2010a; Lukasova i wsp., 2011; Tunaru i wsp., 2003). Podczas gdy niacyna wzmaga odwrócony transport cholesterolu w makrofagach i zmniejsza zmiany miażdżycowe (Lukasova i wsp., 2011), wpływ ?OHB na zmiany miażdżycowe pozostaje nieznany. Chociaż receptor GPR109A pełni role ochronne i istnieją intrygujące powiązania między stosowaniem diety ketogenicznej w udarze a chorobami neurodegeneracyjnymi (Fu i wsp., 2015; Rahman i wsp., 2014), ochronnej roli aOHB za pośrednictwem GPR109A nie wykazano in vivo .
Wreszcie, ?OHB może wpływać na apetyt i sytość. Metaanaliza badań, w których mierzono wpływ diet ketogenicznych i bardzo niskoenergetycznych, wykazała, że uczestnicy spożywający te diety wykazują większe uczucie sytości w porównaniu z dietami kontrolnymi (Gibson i in., 2015). Jednak prawdopodobnym wyjaśnieniem tego efektu są dodatkowe elementy metaboliczne lub hormonalne, które mogą modulować apetyt. Na przykład, myszy utrzymywane na diecie ketogenicznej dla gryzoni wykazywały zwiększone zużycie energii w porównaniu z myszami karmionymi karmą kontrolną, pomimo podobnego spożycia kalorii, a krążąca leptyna lub geny peptydów regulujących zachowanie żywieniowe nie uległy zmianie (Kennedy i wsp., 2007). Wśród proponowanych mechanizmów sugerujących tłumienie apetytu przez ?OHB znajdują się zarówno sygnalizacja, jak i utlenianie (Laeger et al., 2010). Specyficzna dla hepatocytów delecja genu rytmu okołodobowego (Per2) i badania immunoprecypitacji chromatyny wykazały, że PER2 bezpośrednio aktywuje gen Cpt1a i pośrednio reguluje Hmgcs2, prowadząc do upośledzenia ketozy u myszy z nokautem Per2 (Chavan i wsp., 2016). Myszy te wykazywały upośledzone przewidywanie pokarmu, które zostało częściowo przywrócone przez ogólnoustrojowe podawanie ?OHB. Potrzebne będą dalsze badania, aby potwierdzić, że ośrodkowy układ nerwowy jest bezpośrednim celem ?OHB i czy do zaobserwowanych efektów wymagane jest utlenianie ketonów, czy też zaangażowany jest inny mechanizm sygnalizacji. Inni badacze powoływali się na możliwość miejscowej ketogenezy pochodzącej z astrocytów w brzuszno-przyśrodkowym podwzgórzu jako regulatora przyjmowania pokarmu, ale te wstępne obserwacje również skorzystają na ocenach genetycznych i opartych na przepływie (Le Foll i wsp., 2014). Związek między ketozą a niedoborem składników odżywczych pozostaje interesujący, ponieważ głód i sytość są ważnymi elementami nieudanych prób odchudzania.
Integracja metabolizmu ciała ketonowego, modyfikacji potranslacyjnej i fizjologii komórki
Ciała ketonowe przyczyniają się do skompartmentalizowanej puli acetylo-CoA, kluczowego związku pośredniego, który odgrywa znaczącą rolę w metabolizmie komórkowym (Pietrocola i wsp., 2015). Jedną z funkcji acetylo-CoA jest służenie jako substrat do acetylacji, katalizowanej enzymatycznie modyfikacji kowalencyjnej histonu (Choudhary i wsp., 2014; Dutta i wsp., 2016; Fan i wsp., 2015; Menzies i wsp., 2016 ). Duża liczba dynamicznie acetylowanych białek mitochondrialnych, z których wiele może powstać w wyniku mechanizmów nieenzymatycznych, również wyłoniła się z badań proteomiki obliczeniowej (Dittenhafer-Reed i wsp., 2015; Hebert i wsp., 2013; Rardin i wsp., 2013 ; Shimazu i wsp., 2010). Deacetylazy lizynowe wykorzystują kofaktor cynkowy (np. nukleocytozolowe HDAC) lub NAD+ jako kosubstrat (sirtuiny, SIRT) (Choudhary i wsp., 2014; Menzies i wsp., 2016). Acetyloproteom służy zarówno jako czujnik, jak i efektor całkowitej komórkowej puli acetylo-CoA, ponieważ każda z manipulacji fizjologicznych i genetycznych powoduje nieenzymatyczne globalne zmiany acetylacji (Weinert et al., 2014). Ponieważ metabolity wewnątrzkomórkowe służą jako modulatory acetylacji reszt lizynowych, ważne jest rozważenie roli ciał ketonowych, których liczebność jest wysoce dynamiczna.
• OHB jest epigenetycznym modyfikatorem działającym na co najmniej dwa mechanizmy. Zwiększone poziomy OHB wywołane postem, ograniczeniem kalorii, bezpośrednim podawaniem lub długotrwałym wysiłkiem fizycznym wywołują hamowanie HDAC lub aktywację acetylotransferazy histonów (Marosi i wsp., 2016; Sleiman i wsp., 2016) lub stres oksydacyjny (Shimazu i wsp., 2013) . • Hamowanie HDAC3 przez OHB może regulować fizjologię metaboliczną noworodka (Rando i wsp., 2016). Niezależnie, sam aOHB bezpośrednio modyfikuje reszty lizyny histonu (Xie i wsp., 2016). Długotrwałe głodzenie lub cukrzycowa kwasica ketonowa wywołana steptozotocyną zwiększały a-hydroksybutyrylację histonu. Chociaż liczba miejsc a-hydroksybutyrylowania lizyny i miejsc acetylacji była porównywalna, zaobserwowano stechiometrycznie większe a-hydroksybutyrylowanie histonu niż acetylowanie. P-hydroksybutyrylacja histonu lizyny w porównaniu z acetylacją lub metylacją miała wpływ na różne geny, co sugeruje odmienne funkcje komórkowe. Nie wiadomo, czy a-hydroksybutyrylacja jest spontaniczna czy enzymatyczna, ale rozszerza zakres mechanizmów poprzez ciałka ketonowe dynamicznie wpływające na transkrypcję.
Niezbędne zdarzenia przeprogramowania komórek podczas restrykcji kalorycznej i niedoboru składników odżywczych mogą zachodzić w zależnej od SIRT3 i SIRT5 deacetylacji i desuccynylacji mitochondriów, odpowiednio, regulując białka ketogenne i ketolityczne na poziomie potranslacyjnym w wątrobie i tkankach pozawątrobowych (Dittenhafer-Reed et al., 2015; Hebert i in., 2013; Rardin i in., 2013; Shimazu i in., 2010). Chociaż porównanie stechiometryczne zajętych miejsc niekoniecznie wiąże się bezpośrednio ze zmianami w przepływie metabolicznym, acetylacja mitochondrialna jest dynamiczna i może być napędzana przez stężenie acetylo-CoA lub pH mitochondrialne, a nie enzymatyczne acetylotransferazy (Wagner i Payne, 2013). Fakt, że SIRT3 i SIRT5 modulują aktywność enzymów metabolizujących ciało ketonowe, nasuwa pytanie o wzajemną rolę ketonów w rzeźbieniu acetylproteomu, sukcynyloproteomu i innych dynamicznych celów komórkowych. Rzeczywiście, ponieważ zmiany ketogenezy odzwierciedlają stężenia NAD +, produkcja i obfitość ketonów mogą regulować aktywność sirtuiny, wpływając w ten sposób na całkowite pule acetylo-CoA / sukcynylo-CoA, acylproteom, a tym samym fizjologię mitochondrialną i komórkową. P-hydroksybutyrylacja reszt enzymu lizyny może dodać kolejną warstwę do przeprogramowania komórkowego. W tkankach pozawątrobowych utlenianie ciał ketonowych może stymulować analogiczne zmiany w homeostazie komórkowej. Podczas gdy podział puli acetylo-CoA jest silnie regulowany i koordynuje szerokie spektrum zmian komórkowych, zdolność ciał ketonowych do bezpośredniego kształtowania zarówno mitochondrialnych, jak i cytoplazmatycznych stężeń acetylo-CoA wymaga wyjaśnienia (Chen et al., 2012; Corbet et al., 2016; Pougovkina i in., 2014; Schwer i in., 2009; Wellen i Thompson, 2012). Ponieważ stężenia acetylo-CoA są ściśle regulowane, a acetylo-CoA jest nieprzepuszczalny dla błony, ważne jest, aby wziąć pod uwagę mechanizmy napędowe koordynujące homeostazę acetylo-CoA, w tym szybkość produkcji i końcowego utleniania w cyklu TCA, konwersja do ciał ketonowych, mitochondrialne wypływ przez acetylotransferazę karnityny (CrAT) lub eksport acetylo-CoA do cytozolu po konwersji do cytrynianu i uwolnieniu przez liazę cytrynianową ATP (ACLY). Kluczowe role tych ostatnich mechanizmów w acetylproteomie komórkowym i homeostazie wymagają odpowiedniego zrozumienia roli ketogenezy i utleniania ketonów (Das i wsp., 2015; McDonnell i wsp., 2016; Moussaieff i wsp., 2015; Overmyer i wsp., 2015; Seiler i in., 2014; Seiler i in., 2015; Wellen i in., 2009; Wellen i Thompson, 2012). Aby określić cele i wyniki, potrzebne będą konwergentne technologie w metabolomice i acylopoteomice w tworzeniu modeli modyfikowanych genetycznie.
Przeciwzapalne i przeciwzapalne reakcje na ciała ketonowe
Ketoza i ciała ketonowe modulują stan zapalny i funkcję komórek odpornościowych, ale zaproponowano różne, a nawet rozbieżne mechanizmy. Długotrwała deprywacja składników odżywczych zmniejsza stan zapalny (Youm i in., 2015), ale przewlekła ketoza cukrzycy typu 1 jest stanem prozapalnym (Jain i wsp., 2002; Kanikarla-Marie i Jain, 2015; Kurepa i wsp., 2012 ). Role sygnalizacyjne oparte na mechanizmie dla OHB w zapaleniu pojawiają się, ponieważ wiele komórek układu odpornościowego, w tym makrofagi lub monocyty, obficie eksprymuje GPR109A. Podczas gdy OHB wywiera głównie odpowiedź przeciwzapalną (Fu i wsp., 2014; Gambhir i wsp., 2012; Rahman i wsp., 2014; Youm i wsp., 2015), wysokie stężenia ciał ketonowych, zwłaszcza AcAc, mogą wywołać reakcję prozapalną (Jain i in., 2002; Kanikarla-Marie i Jain, 2015; Kurepa i in., 2012).
Dokonano przeglądu przeciwzapalnej roli ligandów GPR109A w miażdżycy, otyłości, zapalnej chorobie jelit, chorobie neurologicznej i raku (Graff et al., 2016). Ekspresja GPR109A jest zwiększona w komórkach RPE modeli cukrzycowych, ludzi z cukrzycą (Gambhir et al., 2012) oraz w mikrogleju podczas neurodegeneracji (Fu et al., 2014). Efekty przeciwzapalne aOHB są wzmacniane przez nadekspresję GPR109A w komórkach RPE i niwelowane przez farmakologiczne hamowanie lub genetyczny knockout GPR109A (Gambhir i wsp., 2012). ?OHB i egzogenny kwas nikotynowy (Taggart i wsp., 2005), oba nadają działanie przeciwzapalne w TNF? lub stan zapalny wywołany przez LPS przez zmniejszenie poziomu białek prozapalnych (iNOS, COX-2) lub wydzielanych cytokin (TNFa, IL-1a, IL-6, CCL2/MCP-1), częściowo poprzez hamowanie NF Translokacja -?B (Fu i wsp., 2014; Gambhir i wsp., 2012). ?OHB zmniejsza stres ER i inflamasom NLRP3, aktywując odpowiedź na stres antyoksydacyjny (Bae i wsp., 2016; Youm i wsp., 2015). Jednak w zapaleniu neurodegeneracyjnym, ochrona zależna od GPR109A, w której pośredniczy ?OHB, nie obejmuje mediatorów zapalnych, takich jak sygnalizacja szlaku MAPK (np. ERK, JNK, p38) (Fu i wsp., 2014), ale może wymagać PGD1 zależnej od COX-2 produkcja (Rahman i in., 2014). Intrygujące jest to, że makrofag GPR109A jest wymagany do wywierania działania neuroprotekcyjnego w modelu udaru niedokrwiennego (Rahman et al., 2014), ale zdolność ?OHB do hamowania inflamasomu NLRP3 w makrofagach pochodzących ze szpiku kostnego jest niezależna od GPR109A (Youm et al. ., 2015). Chociaż większość badań łączy ?OHB z działaniem przeciwzapalnym, ?OHB może mieć działanie prozapalne i zwiększać markery peroksydacji lipidów w hepatocytach cielęcych (Shi i wsp., 2014). Efekty przeciwzapalne lub prozapalne aOHB mogą zatem zależeć od typu komórki, stężenia aOHB, czasu ekspozycji oraz obecności lub braku komodulatorów.
W przeciwieństwie do OHB, AcAc może aktywować sygnalizację prozapalną. Podwyższony AcAc, szczególnie przy wysokim stężeniu glukozy, nasila uszkodzenie komórek śródbłonka poprzez mechanizm zależny od oksydazy NADPH / stresu oksydacyjnego (Kanikarla-Marie i Jain, 2015). Wysokie stężenia AcAc w pępowinie matek z cukrzycą były skorelowane z wyższym tempem utleniania białek i stężeniem MCP-1 (Kurepa i in., 2012). Wysoki poziom AcAc u pacjentów z cukrzycą był skorelowany z TNF? ekspresja (Jain i wsp., 2002) i AcAc, ale nie OHB, indukowały TNFa, ekspresję MCP-1, akumulację ROS i obniżony poziom cAMP w ludzkich komórkach monocytów U937 (Jain i wsp., 2002; Kurepa i wsp. ., 2012).
Zjawiska sygnalizacyjne zależne od ciał ketonowych są często wyzwalane jedynie przy wysokich stężeniach ciał ketonowych (> 5 mM), aw przypadku wielu badań łączących ketony z działaniem pro lub przeciwzapalnym, poprzez niejasne mechanizmy. Ponadto, ze względu na sprzeczny wpływ OHB w porównaniu z AcAc na stan zapalny oraz zdolność stosunku AcAc / OHB do wpływania na mitochondrialny potencjał redoks, najlepsze eksperymenty oceniające rolę ciał ketonowych w fenotypach komórkowych porównują wpływ AcAc i? OHB w różnych stosunkach i przy różnych skumulowanych stężeniach [np. (Saito i in., 2016)]. Wreszcie AcAc można kupić na rynku tylko jako sól litu lub jako ester etylowy, który przed użyciem wymaga hydrolizy zasadowej. Kation litu niezależnie indukuje kaskady transdukcji sygnału (Manji i wsp., 1995), a anion AcAc jest labilny. Wreszcie, badania z użyciem racemicznego d / l-a-OHB mogą być skomplikowane, ponieważ tylko stereoizomer d-a-OHB może być utleniony do AcAc, ale każdy z d-a-OHB i l-a-OHB może sygnalizować przez GPR109A, hamować inflamasom NLRP3, i służą jako substraty lipogenne.
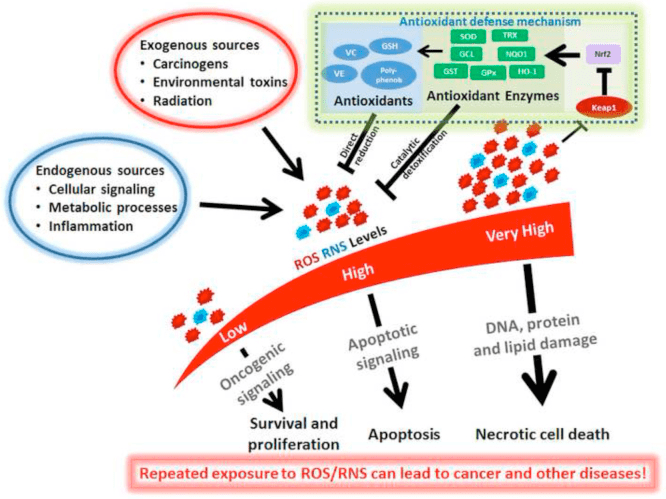
Ketone Bodies, Oxidative Stress i Neuroprotection
Stres oksydacyjny jest zwykle definiowany jako stan, w którym ROS występują w nadmiarze z powodu nadmiernej produkcji i/lub upośledzonej eliminacji. Rola ciał ketonowych jako przeciwutleniająca i łagodząca stres oksydacyjny została szeroko opisana zarówno in vitro, jak i in vivo, szczególnie w kontekście neuroprotekcji. Ponieważ większość neuronów nie wytwarza skutecznie fosforanów o wysokiej energii z kwasów tłuszczowych, ale utlenia ciała ketonowe, gdy brakuje węglowodanów, neuroprotekcyjne działanie ciał ketonowych jest szczególnie ważne (Cahill GF Jr, 2006; Edmond i in., 1987; Yang i in., 1987). W modelach stresu oksydacyjnego indukcja BDH1 i supresja SCOT sugerują, że metabolizm ciała ketonowego można przeprogramować, aby podtrzymać różne sygnalizacje komórkowe, potencjał redoks lub wymagania metaboliczne (Nagao i wsp., 2016; Tieu i wsp., 2003).
Ciała ketonowe zmniejszają stopień uszkodzenia komórek, urazów, śmierci i niższej apoptozy w neuronach i kardiomiocytach (Haces i wsp., 2008; Maalouf i wsp., 2007; Nagao i wsp., 2016; Tieu i wsp., 2003). Wywoływane mechanizmy są zróżnicowane i nie zawsze są liniowo powiązane z koncentracją. Niskie milimolowe stężenia (d lub l) - A OHB wychwytujący RFT (anion hydroksylowy), podczas gdy AcAc zmiata wiele gatunków RFT, ale tylko w stężeniach przekraczających fizjologiczny zakres (IC50 20 mM) (Haces i in., 67) . I odwrotnie, korzystny wpływ na potencjał redoks łańcucha transportu elektronów jest mechanizmem powszechnie powiązanym z d-aOHB. Podczas gdy wszystkie trzy ciała ketonowe (d / l-aOHB i AcAc) zmniejszyły śmierć komórek neuronalnych i akumulację RFT wyzwalane przez chemiczne hamowanie glikolizy, tylko d-a-OHB i AcAc zapobiegały spadkowi neuronalnego ATP. Odwrotnie, w modelu hipoglikemii in vivo, (d lub l) -? OHB, ale nie AcAc zapobiegał peroksydacji lipidów w hipokampie (Haces i in., 2008; Maalouf i in., 2008; Marosi i in., 2007; Murphy, 2016 ; Tieu i in., 2009). Badania in vivo myszy karmionych dietą ketogenną (2003% tłuszczu kcal i 87% białka) wykazały neuroanatomiczne zróżnicowanie zdolności antyoksydacyjnej (Ziegler et al., 13), gdzie najgłębsze zmiany obserwowano w hipokampie, ze wzrostem peroksydazy glutationowej i całkowitej zdolności przeciwutleniające.
Dieta ketogeniczna, estry ketonowe (patrz także Terapeutyczne zastosowanie diety ketogenicznej i egzogennych ciał ketonowych) lub podawanie OHB wywiera działanie neuroprotekcyjne w modelach udaru niedokrwiennego (Rahman i wsp., 2014); Choroba Parkinsona (Tieu i wsp., 2003); napad toksyczności tlenowej ośrodkowego układu nerwowego (D'Agostino i wsp., 2013); skurcze epileptyczne (Yum i wsp., 2015); encefalomiopatia mitochondrialna, zespół kwasicy mleczanowej i epizodów podobnych do udaru mózgu (MELAS) (Frey i wsp., 2016) oraz choroba Alzheimera (Cunnane i Crawford, 2003; Yin i wsp., 2016). Z kolei niedawny raport wykazał histopatologiczne dowody na progresję neurodegeneracyjną spowodowaną dietą ketogenną w transgenicznym mysim modelu nieprawidłowej naprawy mitochondrialnego DNA, pomimo wzrostu biogenezy mitochondrialnej i sygnatur antyoksydacyjnych (Lauritzen i wsp., 2016). Inne sprzeczne raporty sugerują, że ekspozycja na wysokie stężenia ciał ketonowych wywołuje stres oksydacyjny. Wysokie dawki OHB lub AcAc indukowały wydzielanie tlenku azotu, peroksydację lipidów, zmniejszoną ekspresję SOD, peroksydazy glutationowej i katalazy w hepatocytach cielęcych, podczas gdy w hepatocytach szczurów indukcję szlaku MAPK przypisywano AcAc, ale nie OHB (Abdelmegeed et al., 2004 ; Shi i in., 2014; Shi i in., 2016).
Podsumowując, większość doniesień łączy OHB z osłabieniem stresu oksydacyjnego, ponieważ jego podawanie hamuje wytwarzanie ROS / nadtlenków, zapobiega peroksydacji lipidów i utlenianiu białek, zwiększa poziom białek przeciwutleniających oraz poprawia oddychanie mitochondrialne i produkcję ATP (Abdelmegeed i wsp., 2004; Haces i wsp., 2008; Jain i wsp., 1998; Jain i wsp., 2002; Kanikarla-Marie i Jain, 2015; Maalouf i wsp., 2007; Maalouf i Rho, 2008; Marosi i wsp., 2016; Tieu i in., 2003; Yin i in., 2016; Ziegler i in., 2003). Chociaż AcAc był bardziej bezpośrednio skorelowany niż AOHB z indukcją stresu oksydacyjnego, efekty te nie zawsze są łatwo oddzielone od potencjalnych odpowiedzi prozapalnych (Jain i wsp., 2002; Kanikarla-Marie i Jain, 2015; Kanikarla-Marie i Jain, 2016). Ponadto należy wziąć pod uwagę, że pozorna korzyść antyoksydacyjna zapewniana przez plejotropową dietę ketogenną może nie być transdukowana przez same ciała ketonowe, a neuroprotekcja zapewniana przez ciałka ketonowe może nie być całkowicie przypisana stresowi oksydacyjnemu. Na przykład podczas deprywacji glukozy, w modelu deprywacji glukozy w neuronach korowych, aOHB stymulował przepływ autofagiczny i zapobiegał akumulacji autofagosomów, co było związane ze zmniejszeniem śmierci neuronów (Camberos-Luna i in., 2016). d-aOHB indukuje również kanoniczne białka przeciwutleniające FOXO3a, SOD, MnSOD i katalazę, prospektywnie poprzez hamowanie HDAC (Nagao i wsp., 2016; Shimazu i wsp., 2013).
Niealkoholowa stłuszczenie wątroby (NAFLD) i metabolizm ciała ketonowego
NAFLD związane z otyłością i niealkoholowe stłuszczeniowe zapalenie wątroby (NASH) są najczęstszymi przyczynami chorób wątroby w krajach zachodnich (Rinella i Sanyal, 2016), a niewydolność wątroby wywołana przez NASH jest jedną z najczęstszych przyczyn przeszczepu wątroby. Podczas gdy samo nadmierne przechowywanie triacylogliceroli w hepatocytach >5% masy wątroby (NAFL) nie powoduje degeneracyjnej czynności wątroby, progresja do NAFLD u ludzi koreluje z ogólnoustrojową opornością na insulinę i zwiększonym ryzykiem cukrzycy typu 2 i może przyczyniać się do patogenezy choroba sercowo-naczyniowa i przewlekła choroba nerek (Fabbrini i wsp., 2009; Targher i wsp., 2010; Targher i Byrne, 2013). Mechanizmy patogenne NAFLD i NASH nie są w pełni poznane, ale obejmują nieprawidłowości w metabolizmie hepatocytów, autofagię hepatocytów i stres retikulum endoplazmatycznego, funkcję wątrobowych komórek odpornościowych, zapalenie tkanki tłuszczowej i ogólnoustrojowe mediatory zapalne (Fabbrini i wsp., 2009; Masuoka i Chalasani, 2013 Targher i wsp., 2010; Yang i wsp., 2010). Zaburzenia metabolizmu węglowodanów, lipidów i aminokwasów występują i przyczyniają się do otyłości, cukrzycy i NAFLD u ludzi i organizmów modelowych [przegląd w (Farese i in., 2012; Lin i Accili, 2011; Newgard, 2012; Samuel i Shulman, 2012; Sun i Lazar, 2013)]. Podczas gdy nieprawidłowości hepatocytów w cytoplazmatycznym metabolizmie lipidów są powszechnie obserwowane w NAFLD (Fabbrini i wsp., 2010b), rola metabolizmu mitochondrialnego, który reguluje oksydacyjną utylizację tłuszczów, jest mniej jasna w patogenezie NAFLD. Nieprawidłowości metabolizmu mitochondrialnego występują i przyczyniają się do patogenezy NAFLD/NASH (Hyotylainen i wsp., 2016; Serviddio i wsp., 2011; Serviddio i wsp., 2008; Wei i wsp., 2008). Są ogólne (Felig et al., 1974; Iozzo et al., 2010; Koliaki et al., 2015; Satapati et al., 2015; Satapati et al., 2012; Sunny et al., 2011), ale nie jednolite ( Koliaki i Roden, 2013; Perry i wsp., 2016; Rector i wsp., 2010) zgadzają się, że przed rozwojem NASH w dobrej wierze, wątrobowe utlenianie mitochondriów, a w szczególności utlenianie tłuszczów, nasila się w otyłości, ogólnoustrojowej oporności na insulinę i NAFLD. Jest prawdopodobne, że wraz z postępem NAFLD pojawia się heterogenność zdolności oksydacyjnej, nawet wśród poszczególnych mitochondriów, i ostatecznie funkcja oksydacyjna zostaje osłabiona (Koliaki i in., 2015; Rector i in., 2010; Satapati i in., 2008; Satapati i in., 2012; ., XNUMX).
Ketogeneza jest często używana jako zastępca utleniania tłuszczu w wątrobie. Zaburzenia ketogenezy pojawiają się wraz z postępem NAFLD w modelach zwierzęcych i prawdopodobnie u ludzi. Poprzez nie do końca zdefiniowane mechanizmy hiperinsulinemia hamuje ketogenezę, prawdopodobnie przyczyniając się do hipoketonemii w porównaniu do szczupłych grup kontrolnych (Bergman i wsp., 2007; Bickerton i wsp., 2008; Satapati i wsp., 2012; Soeters i wsp., 2009; Sunny i wsp., 2011; Bickerton i wsp., 2005; Satapati i wsp., 2015; Soeters i wsp., 2001; Sunny i wsp. , 2012; Vice i in., 2010). Niemniej jednak zdolność krążących ciał ketonowych do przewidywania NAFLD jest kontrowersyjna (M nnist i in., 2008; Sanyal i in., 2011). Solidne ilościowe metody spektroskopowe rezonansu magnetycznego w modelach zwierzęcych ujawniły zwiększoną szybkość obrotu ketonowego przy umiarkowanej insulinooporności, ale niższe wskaźniki były widoczne przy cięższej insulinooporności (Satapati i in., 4; Sunny i in., 2015). U otyłych ludzi ze stłuszczeniem wątroby szybkość ketogenezy jest normalna (Bickerton i in., 2012; Sunny i in., 2015), a zatem tempo ketogenezy jest zmniejszone w stosunku do zwiększonego obciążenia kwasami tłuszczowymi w hepatocytach. W konsekwencji acetylo-CoA pochodzący z a-oksydacji może być skierowany na końcowe utlenianie w cyklu TCA, zwiększając końcowe utlenianie, glukoneogenezę wywoływaną przez fosfoenolopirogronian poprzez anaplerozę / kataplerozę i stres oksydacyjny. Acetylo-CoA prawdopodobnie jest również eksportowany z mitochondriów jako cytrynian, prekursorowy substrat lipogenezy (ryc. 2012) (Satapati i in., 1; Satapati i in., 2016; Solinas i in., 1). Podczas gdy ketogeneza staje się mniej wrażliwa na insulinę lub post z przedłużającą się otyłością (Satapati i wsp., 2), podstawowe mechanizmy i dalsze konsekwencje tego pozostają nie w pełni zrozumiałe. Niedawne dowody wskazują, że mTORC2010 hamuje ketogenezę w sposób, który może następować za sygnalizacją insuliny (Kucejova i wsp., 2), co jest zgodne z obserwacjami, że mTORC1 hamuje indukcję HmgcsXNUMX pośredniczoną przez PPARy (Sengupta i wsp., XNUMX) ( zobacz także rozporządzenie HMGCSXNUMX i SCOT / OXCTXNUMX).

Wstępne obserwacje z naszej grupy sugerują niekorzystne następstwa niewydolności ketogennej dla wątroby (Cotter et al., 2014). Aby przetestować hipotezę, że upośledzona ketogeneza, nawet w stanach bogatych w węglowodany, a tym samym `` nieketogennych '', przyczynia się do nieprawidłowego metabolizmu glukozy i wywołuje stłuszczeniowe zapalenie wątroby, stworzyliśmy mysi model znacznej niewydolności ketogennej poprzez podanie antysensownych oligonukleotydów (ASO) ukierunkowanych na Hmgcs2. Utrata HMGCS2 u standardowych dorosłych myszy karmionych niskotłuszczową karmą spowodowała łagodną hiperglikemię i znacznie zwiększoną produkcję setek metabolitów wątrobowych, z których zestaw silnie sugerował aktywację lipogenezy. Karmienie myszy dietą wysokotłuszczową z niewystarczającą ketogenezą spowodowało rozległe uszkodzenie hepatocytów i stan zapalny. Odkrycia te potwierdzają główne hipotezy, że (i) ketogeneza nie jest pasywną ścieżką przepełnienia, ale raczej dynamicznym węzłem w wątrobowej i zintegrowanej fizjologicznej homeostazie, oraz (ii) rozważne wzmocnienie ketogeniczne w celu złagodzenia NAFLD / NASH i zaburzonego metabolizmu glukozy w wątrobie jest warte zbadania .
W jaki sposób zaburzona ketogeneza może przyczynić się do uszkodzenia wątroby i zmiany homeostazy glukozy? Pierwszą kwestią jest to, czy winowajcą jest niedobór strumienia ketogenicznego, czy też same ketony. Niedawny raport sugeruje, że ciała ketonowe mogą łagodzić uszkodzenie wątroby wywołane stresem oksydacyjnym w odpowiedzi na wielonienasycone kwasy tłuszczowe n-3 (Pawlak i in., 2015). Przypomnijmy, że ze względu na brak ekspresji SCOT w hepatocytach, ciała ketonowe nie są utleniane, ale mogą przyczyniać się do lipogenezy i pełnić różne funkcje sygnalizacyjne niezależne od ich utleniania (patrz także Nieoksydacyjne losy metaboliczne ciał ketonowych i OHB jako mediator sygnałowy). Możliwe jest również, że ciałka ketonowe pochodzące z hepatocytów mogą służyć jako sygnał i / lub metabolit dla sąsiednich typów komórek w obrębie wątroby trądzikowej, w tym komórek gwiaździstych i makrofagów komórek Kupffera. Chociaż dostępna ograniczona literatura sugeruje, że makrofagi nie są zdolne do utleniania ciał ketonowych, zostało to zmierzone tylko przy użyciu klasycznych metod i tylko w makrofagach otrzewnowych (Newsholme i in., 1986; Newsholme i in., 1987), co wskazuje, że re- ocena jest właściwa, biorąc pod uwagę obfitą ekspresję SCOT w makrofagach pochodzących ze szpiku kostnego (Youm et al., 2015).
Strumień ketogenny hepatocytów może również działać cytoprotekcyjnie. Chociaż zbawienne mechanizmy mogą nie zależeć od ketogenezy jako takiej, diety ketogenne o niskiej zawartości węglowodanów są związane z łagodzeniem NAFLD (Browning i wsp., 2011; Foster i wsp., 2010; Kani i wsp., 2014; Schugar i Crawford, 2012) . Nasze obserwacje wskazują, że ketogeneza hepatocytów może powodować sprzężenie zwrotne i regulować przepływ cyklu TCA, strumień anaplerotyczny, glukoneogenezę pochodzącą z fosfoenolopirogronianu (Cotter et al., 2014), a nawet przemianę glikogenu. Upośledzenie ketogeniczne kieruje acetylo-CoA w celu zwiększenia przepływu TCA, co w wątrobie jest powiązane ze zwiększonym uszkodzeniem, w którym pośredniczą ROS (Satapati i in., 2015; Satapati i in., 2012); wymusza przekierowanie węgla na zsyntetyzowane de novo gatunki lipidów, które mogą okazać się cytotoksyczne; i zapobiega ponownemu utlenianiu NADH do NAD + (Cotter i wsp., 2014) (Rys. 4). Podsumowując, przyszłe eksperymenty są potrzebne, aby zająć się mechanizmami, przez które względna niewydolność ketogeniczna może stać się nieprzystosowana, przyczynić się do hiperglikemii, wywołać stłuszczeniowe zapalenie wątroby oraz sprawdzić, czy mechanizmy te działają w ludzkim NAFLD / NASH. Ponieważ dowody epidemiologiczne wskazują na upośledzoną ketogenezę podczas progresji stłuszczeniowego zapalenia wątroby (Embade i wsp., 2016; Marinou i wsp., 2011; M nnist i wsp., 2015; Pramfalk i wsp., 2015; Safaei i wsp., 2016) terapie zwiększające ketogenezę wątrobową mogą okazać się zbawienne (Degirolamo i in., 2016; Honda i in., 2016).
Ciała ketonowe i niewydolność serca (HF)
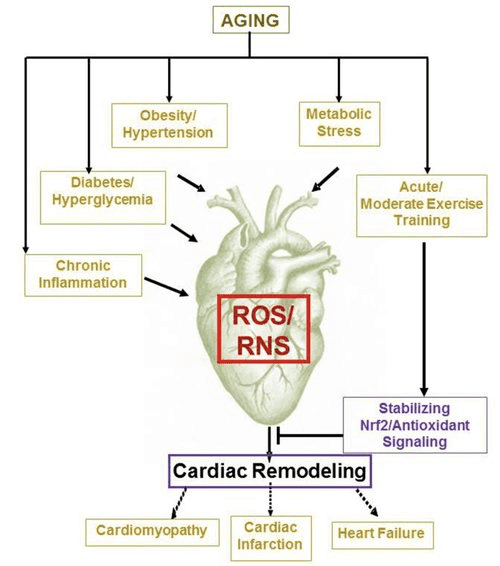
Przy tempie metabolizmu przekraczającym 400 kcal/kg/dzień i obrocie 6 kg ATP/dzień serce jest organem o największym wydatku energetycznym i zapotrzebowaniu oksydacyjnym (Ashrafian i wsp., 35; Wang i wsp., 2007b). Zdecydowana większość obrotu energetycznego mięśnia sercowego znajduje się w mitochondriach, a 2010% tej podaży pochodzi z FAO. Serce jest wszystkożerne i elastyczne w normalnych warunkach, ale serce ulegające patologicznej przebudowie (np. z powodu nadciśnienia lub zawału mięśnia sercowego) i serce z cukrzycą stają się metabolicznie nieelastyczne (Balasse i Fery, 70; BING, 1989; Fukao i in., 1954 Lopaschuk i wsp., 2004; Taegtmeyer i wsp., 2010; Taegtmeyer i wsp., 1980; Young i wsp., 2002). Rzeczywiście, genetycznie zaprogramowane nieprawidłowości metabolizmu paliwa sercowego w modelach mysich wywołują kardiomiopatię (Carley i wsp., 2002; Neubauer, 2014). W warunkach fizjologicznych normalne serce utlenia ciała ketonowe proporcjonalnie do ich dostarczenia, kosztem utleniania kwasów tłuszczowych i glukozy, a mięsień sercowy jest największym konsumentem ciał ketonowych na jednostkę masy (BING, 2007; Crawford i in., 1954; GARLAND i in. 2009; Hasselbaink i wsp. 1962; Jeffrey i wsp. 2003; Pelletier i wsp. 1995; Tardif i wsp. 2007; Yan i wsp. 2001). W porównaniu z utlenianiem kwasów tłuszczowych, ciała ketonowe są bardziej wydajne energetycznie, dostarczając więcej energii dostępnej do syntezy ATP na cząsteczkę zainwestowanego tlenu (stosunek P/O) (Kashiwaya et al., 2009; Sato et al., 2010; Veech, 1995) . Utlenianie ciała ketonowego daje również potencjalnie wyższą energię niż FAO, utrzymując utlenianie ubichinonu, co zwiększa zakres redoks w łańcuchu transportu elektronów i udostępnia więcej energii do syntezy ATP (Sato i wsp., 2004; Veech, 1995). Utlenianie ciał ketonowych może również ograniczać produkcję ROS, a tym samym stres oksydacyjny (Veech, 2004).
Wstępne badania interwencyjne i obserwacyjne wskazują na potencjalną zbawienną rolę ciał ketonowych w sercu. W doświadczalnym kontekście uszkodzenia niedokrwienno-reperfuzyjnego ciałka ketonowe nadają potencjalne działanie kardioprotekcyjne (Al-Zaid i wsp., 2007, Wang i wsp., 2008), prawdopodobnie z powodu zwiększonej ilości mitochondrialnej w sercu lub regulacji w górę kluczowej fosforylacji oksydacyjnej mediatory (Snorek i wsp., 2012; Zou i wsp., 2002). Ostatnie badania wskazują, że wykorzystanie ciała ketonowego wzrasta w wadach serc myszy (Aubert i wsp., 2016) i ludzi (Bedi i wsp., 2016), wspierając wcześniejsze obserwacje u ludzi (BING, 1954, Fukao i wsp., 2000; Janardhan i wsp., 2011, Longo i wsp., 2004, Rudolph i Schinz, 1973, Tildon i Cornblath, 1972). Stężenia ketonów w cyrkulujących ciałach są zwiększone u pacjentów z niewydolnością serca, w bezpośredniej proporcji do ciśnień napełniania, obserwacje, których mechanizm i znaczenie pozostają nieznane (Kupari i wsp., 1995, Lommi i wsp., 1996, Lommi i wsp., 1997, Neely i in. 1972), ale myszy z selektywnym niedoborem SCOT w kardiomiocytach wykazują przyspieszoną patologiczną remodelację komorową i sygnatury ROS w odpowiedzi na chirurgicznie wywołane przeciążenie ciśnieniowe (Schugar i wsp., 2014).
Ostatnie intrygujące obserwacje w terapii cukrzycy ujawniły potencjalny związek między metabolizmem ketonowym mięśnia sercowego a patologiczną przebudową komory (Ryc. 5). Zahamowanie pobranego przez nerkowy proksymalny cylindryczny transporter sodu / glukozy 2 (SGLT2i) zwiększa stężenie w krążeniu ketonów u ludzi (Ferrannini i wsp., 2016a, Inagaki i wsp., 2015) i myszy (Suzuki i wsp., 2014) poprzez zwiększenie ketogeneza wątrobowa (Ferrannini i wsp., 2014, Ferrannini i wsp., 2016a, Katz and Leiter, 2015, Mudaliar i wsp., 2015). Co uderzające, co najmniej jeden z tych środków zmniejszył hospitalizację z powodu HF (np. Jak ujawniono w badaniu EMPA-REG OUTCOME) i zwiększoną śmiertelność z przyczyn sercowo-naczyniowych (Fitchett i wsp., 2016, Sonesson i wsp., 2016, Wu i wsp., 2016a ; Zinman i wsp., 2015). Podczas gdy mechanizmy napędzające korzystne wyniki HF w powiązanym SGLT2i pozostają aktywnie dyskutowane, korzyści z przeżycia są prawdopodobnie wieloczynnikowe, prospektywnie włączając ketozę, ale także zbawienny wpływ na masę ciała, ciśnienie krwi, poziom glukozy i kwasu moczowego, sztywność tętnic, współczulny układ nerwowy, osmotyczny diureza / zmniejszona objętość osocza i zwiększony hematokryt (Raz i Cahn, 2016, Vallon i Thomson, 2016). Podsumowując, przekonanie, że terapeutycznie zwiększający ketonemię u pacjentów z HF lub o wysokim ryzyku rozwoju HF, pozostaje kontrowersyjne, ale jest przedmiotem aktywnych badań w badaniach przedklinicznych i klinicznych (Ferrannini i wsp., 2016b, Kolwicz i wsp., 2016, Lopaschuk i Verma, 2016, Mudaliar i wsp., 2016, Taegtmeyer, 2016).

Ciała ketonowe w biologii raka
Związki między ciałami ketonowymi a rakiem szybko pojawiają się, ale badania zarówno na modelach zwierzęcych, jak i na ludziach przyniosły różne wnioski. Ponieważ metabolizm ketonów jest dynamiczny i reaguje na stan składników odżywczych, kuszące jest utrzymywanie biologicznych powiązań z rakiem, ze względu na potencjał precyzyjnych terapii żywieniowych. Komórki rakowe przechodzą metaboliczne przeprogramowanie w celu utrzymania szybkiej proliferacji i wzrostu komórek (DeNicola i Cantley, 2015, Pavlova i Thompson, 2016). Klasyczny efekt Warburga w metabolizmie komórek rakowych wynika z dominującej roli glikolizy i fermentacji kwasu mlekowego w celu transferu energii i kompensacji mniejszej zależności od oksydacyjnej fosforylacji i ograniczonego oddychania mitochondrialnego (De Feyter i wsp., 2016, Grabacka i wsp., 2016; Kang i wsp., 2015, Poff i wsp., 2014, Shukla i wsp., 2014). Węgiel glukozy jest kierowany głównie przez glikolizę, szlak pentozofosforanowy i lipogenezę, które razem zapewniają związki pośrednie niezbędne do ekspansji biomasy nowotworowej (Grabacka i wsp., 2016, Shukla i wsp., 2014, Yoshii i wsp., 2015). Adaptacja komórek nowotworowych do deprywacji glukozy odbywa się poprzez zdolność do wykorzystywania alternatywnych źródeł paliwa, w tym octanu, glutaminy i asparaginianu (Jaworski i wsp., 2016, Sullivan i wsp., 2015). Na przykład, ograniczony dostęp do pirogronianu ujawnia zdolność komórek rakowych do przekształcania glutaminy w acetylo-CoA przez karboksylację, zachowując zarówno potrzeby energetyczne, jak i anaboliczne (Yang i wsp., 2014). Ciekawą adaptacją komórek rakowych jest zastosowanie octanu jako paliwa (Comerford i wsp., 2014, Jaworski i wsp., 2016, Mashimo i wsp., 2014, Wright i Simone, 2016, Yoshii i wsp., 2015). Octan jest również substratem dla lipogenezy, która jest krytyczna dla proliferacji komórek nowotworowych, a wzrost tego lipogennego kanału jest związany z krótszym przeżyciem pacjenta i większym obciążeniem nowotworowym (Comerford i wsp., 2014, Mashimo i wsp., 2014; Yoshii i in. ., 2015).
Komórki nienowotworowe łatwo przenoszą swoje źródło energii z glukozy do ciał ketonowych podczas niedoboru glukozy. Ta plastyczność może być bardziej zmienna w różnych typach komórek rakowych, ale guzy mózgu wszczepione in vivo utleniają się [2,4-13C2] - aOHB w podobnym stopniu jak otaczająca tkanka mózgowa (De Feyter et al., 2016). Modele `` Odwrotnego efektu Warburga '' lub `` metabolizmu guza dwuprzedziałowego '' zakładają hipotezę, że komórki rakowe indukują produkcję OHB w sąsiednich fibroblastach, zaspokajając potrzeby energetyczne komórek nowotworowych (Bonuccelli et al., 2010; Martinez-Outschoorn et al., 2012) . W wątrobie, przesunięcie hepatocytów od ketogenezy do utleniania ketonów w komórkach raka wątrobowokomórkowego (wątrobiaka) jest zgodne z aktywacją aktywności BDH1 i SCOT obserwowaną w dwóch liniach komórkowych wątrobiaka (Zhang i wsp., 1989). Rzeczywiście, komórki wątrobiaka eksprymują OXCT1 i BDH1 i utleniają ketony, ale tylko wtedy, gdy brakuje surowicy (Huang i wsp., 2016). Alternatywnie zaproponowano również ketogenezę komórek nowotworowych. Dynamiczne zmiany w ekspresji genów ketogennych są widoczne podczas transformacji nowotworowej nabłonka okrężnicy, typu komórek, które normalnie eksprymują HMGCS2, a niedawny raport sugerował, że HMGCS2 może być markerem prognostycznym złego rokowania w raku jelita grubego i płaskonabłonkowego (Camarero i wsp., 2006; Chen i in., 2016). Pozostaje do ustalenia, czy ten związek wymaga ketogenezy, czy też funkcji księżycowej HMGCS2, czy też się z nią wiąże. I odwrotnie, pozorna produkcja αOHB przez komórki czerniaka i glejaka, stymulowana przez PPAR ™. agonista fenofibrat, był związany z zatrzymaniem wzrostu (Grabacka i wsp., 2016). Konieczne są dalsze badania, aby scharakteryzować role ekspresji HMGCS2 / SCOT, ketogenezy i utleniania ketonów w komórkach nowotworowych.
Poza dziedziną metabolizmu paliw, ketony zostały ostatnio zaangażowane w biologię komórek rakowych poprzez mechanizm sygnalizacyjny. Analiza czerniaka BRAF-V600E + wykazała zależną od OCT1 indukcję HMGCL w sposób onkogenny zależny od BRAF (Kang i wsp., 2015). Zwiększenie HMGCL było skorelowane z wyższym stężeniem AcAc w komórkach, co z kolei wzmocniło interakcję BRAFV600E-MEK1, wzmacniając sygnalizację MEK-ERK w pętli sprzężenia zwrotnego, która napędza proliferację i wzrost komórek nowotworowych. Obserwacje te nasuwają intrygujące pytanie o potencjalną pozawątrobową ketogenezę, która następnie wspiera mechanizm sygnalizacji (patrz także? OHB jako mediator sygnalizacji i kontrowersje w pozawątrobowej ketogenezie). Ważne jest również rozważenie niezależnego wpływu AcAc, d-aOHB i l-yOHB na metabolizm raka, a biorąc pod uwagę HMGCL, katabolizm leucyny może być również zaburzony.
Skutki diet ketogenicznych (patrz również Terapeutyczne zastosowanie diety ketogenicznej i egzogennych ciał ketonowych) w modelach zwierzęcych raka są zróżnicowane (De Feyter i in., 2016; Klement i in., 2016; Meidenbauer i in., 2015; Poff i in., 2014 ., 2011; Seyfried i wsp., 2014; Shukla i wsp., 2016). Podczas gdy powiązania epidemiologiczne między otyłością, rakiem i dietami ketogenicznymi są przedmiotem debaty (Liskiewicz i in., 2016; Wright i Simone, 2016), metaanaliza wykorzystująca diety ketogeniczne w modelach zwierzęcych i badaniach na ludziach sugeruje zbawienny wpływ na przeżycie, z korzyści prospektywnie związane z wielkością ketozy, czasem rozpoczęcia diety i lokalizacją guza (Klement i in., 2016; Woolf i in., 81). Leczenie komórek raka trzustki ciałami ketonowymi (d-aOHB lub AcAc) zahamowało wzrost, proliferację i glikolizę, a dieta ketogeniczna (18% kcal tłuszczu, 1% białka, 2014% węglowodanów) zmniejszyła in vivo masę guza, glikemię i wzrost masy mięśniowej i ciała u zwierząt z wszczepionym rakiem (Shukla i wsp., 2014). Podobne wyniki zaobserwowano przy użyciu modelu komórek przerzutowych glejaka u myszy, które otrzymały suplementację ketonową w diecie (Poff i wsp., 91). I odwrotnie, dieta ketogeniczna (9% kcal tłuszczu, 2016% białka) zwiększyła stężenie krążącego ?OHB i zmniejszyła glikemię, ale nie miała wpływu ani na objętość guza, ani na czas przeżycia u szczurów z glejakiem (De Feyter i wsp., 2015). Jako wskaźnik kliniczny, który poprawia zarządzanie metaboliczne w terapii raka mózgu wywołanego dietą ketogenną u ludzi i myszy, zaproponowano indeks glukozowo-ketonowy (Meidenbauer i in., XNUMX). Podsumowując, role metabolizmu ciał ketonowych i ciał ketonowych w biologii raka są kuszące, ponieważ każdy z nich stanowi wykonalne opcje terapeutyczne, ale podstawowe aspekty pozostają do wyjaśnienia, z wyraźnymi wpływami wyłaniającymi się z macierzy zmiennych, w tym (i) różnic między egzogennymi ketonami organizmy a dieta ketogeniczna, (ii) typ komórek rakowych, polimorfizmy genomowe, stopień i stadium zaawansowania; oraz (iii) czas i czas trwania ekspozycji na stan ketotyczny.

Ketogeneza jest tworzona przez ciała ketonowe poprzez rozkład kwasów tłuszczowych i aminokwasów ketogennych. Ten biochemiczny proces dostarcza energii do różnych narządów, szczególnie do mózgu, w warunkach postu jako odpowiedź na niedostępność glukozy we krwi. Ciała ketonowe są produkowane głównie w mitochondriach komórek wątroby. Podczas gdy inne komórki są zdolne do przeprowadzenia ketogenezy, nie są tak skuteczne w robieniu takich komórek jak komórki wątroby. Ponieważ ketogeneza występuje w mitochondriach, jej procesy są regulowane niezależnie. Dr Alex Jimenez DC, CCST Insight
Terapeutyczne zastosowanie diety ketogenicznej i egzogennych ciał ketonowych
Zastosowania diet ketogenicznych i ciał ketonowych jako narzędzi terapeutycznych pojawiły się również w kontekstach nienowotworowych, w tym otyłości i NAFLD/NASH (Browning i in., 2011; Foster i in., 2010; Schugar i Crawford, 2012); niewydolność serca (Huynh, 2016; Kolwicz i in., 2016; Taegtmeyer, 2016); choroby neurologiczne i neurodegeneracyjne (Martin i in., 2016; McNally i Hartman, 2012; Rho, 2015; Rogawski i in., 2016; Yang i Cheng, 2010; Yao i in., 2011); wrodzone błędy metabolizmu (Scholl-B�rgi i in., 2015); i wydajność ćwiczeń (Cox i in., 2016). Skuteczność diet ketogenicznych została szczególnie doceniona w terapii napadów padaczkowych, zwłaszcza u pacjentów lekoopornych. W większości badań oceniano diety ketogeniczne u pacjentów pediatrycznych i wykazano do ~50% zmniejszenie częstości napadów po 3 miesiącach, z lepszą skutecznością w wybranych zespołach (Wu i wsp., 2016b). Doświadczenie jest bardziej ograniczone w przypadku padaczki dorosłych, ale widoczna jest podobna redukcja, z lepszą odpowiedzią u pacjentów z objawową padaczką uogólnioną (Nei et al., 2014). Podstawowe mechanizmy przeciwdrgawkowe pozostają niejasne, chociaż postulowane hipotezy obejmują zmniejszone wykorzystanie glukozy/glikolizę, przeprogramowany transport glutaminianu, pośredni wpływ na wrażliwy na ATP kanał potasowy lub receptor adenozyny A1, zmianę ekspresji izoformy kanału sodowego lub wpływ na krążące hormony, w tym leptynę ( Lambrechts i wsp., 2016; Lin i wsp., 2017; Lutas i Yellen, 2013). Pozostaje niejasne, czy efekt przeciwdrgawkowy można przypisać przede wszystkim ciałkom ketonowym, czy też kaskadowym metabolicznym konsekwencjom diety niskowęglowodanowej. Niemniej jednak estry ketonowe (patrz poniżej) wydają się podnosić próg drgawkowy w zwierzęcych modelach napadów prowokowanych (Ciarlone i in., 2016; D'Agostino i in., 2013; Viggiano i in., 2015).
Dieta w stylu Atkinsa i ketogeniczna dieta o niskiej zawartości węglowodanów są często uważane za nieprzyjemne i mogą powodować zaparcia, hiperurykemię, hipokalcemię, hipomagnezemię, prowadzić do kamicy nerkowej, kwasicy ketonowej, powodować hiperglikemię oraz zwiększać stężenie cholesterolu we krwi i wolnego kwasu tłuszczowego (Bisschop i in., 2001 Kossoff i Hartman, 2012, Kwiterovich i wsp., 2003, Suzuki i wsp., 2002). Z tych powodów długotrwałe stosowanie stawia wyzwania. Badania gryzoni często wykorzystują charakterystyczny rozkład makroskładników (94% kcal, 1% kcal węglowodan, 5% kcal protein, Bio-Serv F3666), który wywołuje silną ketozę. Jednak zwiększenie zawartości białka nawet do 10% kcal znacznie zmniejsza ketozę, a ograniczenie białka 5% kcal wywołuje zakłócające efekty metaboliczne i fizjologiczne. Ten preparat diety jest także pozbawiony choliny, inna zmienna, która wpływa na podatność na uszkodzenie wątroby, a nawet ketogenezę (Garbow i wsp., 2011, Jornayvaz i wsp., 2010, Kennedy i wsp., 2007, Pissios i wsp., 2013; Schugar; i wsp., 2013). Skutki długotrwałego stosowania diet ketogennych u myszy nie zostały jeszcze w pełni określone, ale ostatnie badania na myszach wykazały normalne przeżycie i brak markerów uszkodzenia wątroby u myszy na dietach ketogennych w ciągu ich życia, chociaż metabolizm aminokwasów, wydatek energetyczny i sygnalizacja insulinowa zostały znacznie przeprogramowane (Douris et al., 2015).
Mechanizmy zwiększające ketozę poprzez mechanizmy alternatywne dla diet ketogennych obejmują stosowanie prekursorów ciała ketonowego do spożycia. Podawanie egzogennych ciał ketonowych może stworzyć wyjątkowy stan fizjologiczny, nie spotykany w normalnej fizjologii, ponieważ stężenia glukozy i insuliny w krążeniu są względnie normalne, podczas gdy komórki mogą oszczędzić wychwytu glukozy i jej wykorzystania. Same ciała ketonowe mają krótki okres półtrwania, a spożycie lub wlew soli sodowej a-OHB w celu uzyskania terapeutycznej ketozy wywołuje niepożądane obciążenie sodem. R / S-1,3-butanodiol jest nietoksycznym dialkoholem, który łatwo utlenia się w wątrobie z wytworzeniem d / l-aOHB (Desrochers i wsp., 1992). W różnych kontekstach eksperymentalnych tę dawkę podawano codziennie myszom lub szczurom nawet przez siedem tygodni, uzyskując krążące stężenie OHB do 5 mM w ciągu 2 godzin od podania, które jest stabilne przez co najmniej dodatkowe 3 godziny (D ' Agostino i in., 2013). Częściowe zahamowanie przyjmowania pokarmu obserwowano u gryzoni, którym podawano R / S-1,3-butanodiol (Carpenter and Grossman, 1983). Ponadto trzy chemicznie różne estry ketonowe (KE), (i) monoester R-1,3-butanodiolu i d-a-OHB (R-3-hydroksybutylo-R-a-OHB); (ii) glicerylo-tris-aOHB; oraz (iii) diester acetooctanowy R, S-1,3-butanodiolu, również był szeroko badany (Brunengraber, 1997; Clarke i in., 2012a; Clarke i in., 2012b; Desrochers i in., 1995a; Desrochers i in. ., 1995b; Kashiwaya i in., 2010). Nieodłączną zaletą tego pierwszego jest to, że po hydrolizie esterazy w jelicie lub wątrobie wytwarzane są 2 mole fizjologicznej d-aOHB na mol KE. Bezpieczeństwo, farmakokinetykę i tolerancję najszerzej badano u ludzi przyjmujących R-3-hydroksybutylo R-aOHB w dawkach do 714 mg / kg, uzyskując krążące stężenia d-a-OHB do 6 mM (Clarke et al., 2012a; Cox i in., 2016; Kemper i in., 2015; Shivva i in., 2016). U gryzoni ten KE zmniejsza kaloryczność i całkowity cholesterol w osoczu, stymuluje brązową tkankę tłuszczową i polepsza oporność na insulinę (Kashiwaya i wsp., 2010, Kemper i wsp., 2015, Veech, 2013). Niedawne odkrycia wskazują, że podczas ćwiczeń u wytrenowanych sportowców spożycie R-3-hydroksybutylo R-aOHB zmniejszyło glikolizę mięśni szkieletowych i stężenie mleczanu w osoczu, zwiększyło domięśniowe utlenianie triacyloglicerolu i zachowało zawartość glikogenu w mięśniach, nawet gdy jednocześnie spożywane węglowodany stymulowały wydzielanie insuliny ( Cox i in., 2016). Konieczny jest dalszy rozwój tych intrygujących wyników, ponieważ poprawa wydajności ćwiczeń wytrzymałościowych była głównie spowodowana silną odpowiedzią na KE u pacjentów 2 / 8. Niemniej jednak, wyniki te potwierdzają klasyczne badania wskazujące na preferencję do utleniania ketonów względem innych substratów (GARLAND i wsp., 1962, Hasselbaink i wsp., 2003, Stanley i wsp., 2003, Valente-Silva i wsp., 2015), w tym podczas ćwiczeń, i że wyszkoleni sportowcy mogą być bardziej przygotowani do wykorzystania ketonów (Johnson i wsp., 1969a, Johnson i Walton, 1972, Winder i wsp., 1974, Winder i wsp., 1975). Wreszcie, należy określić mechanizmy, które mogą wspierać poprawę wydajności wysiłkowej po równym spożyciu kalorii (zróżnicowane rozmieszczenie wśród makroelementów) i równe wskaźniki zużycia tlenu.
Przyszłe perspektywy
Niedawno napiętnowane jako ścieżka przelewowa zdolna do akumulacji toksycznych emisji ze spalania tłuszczu w stanach ograniczenia węglowodanów (paradygmat „ketotoksyczny”), ostatnie obserwacje potwierdzają pogląd, że metabolizm ciała ketonowego pełni zbawienną rolę nawet w stanach obciążonych węglowodanami, otwierając „ketohormetykę”. hipoteza. Podczas gdy łatwe podejście żywieniowe i farmakologiczne do manipulowania metabolizmem ketonów czyni go atrakcyjnym celem terapeutycznym, agresywnie nastawione, ale rozważne eksperymenty pozostają w laboratoriach badawczych zarówno podstawowych, jak i translacyjnych. Niezaspokojone potrzeby pojawiły się w dziedzinach definiowania roli dźwigni metabolizmu ketonów w niewydolności serca, otyłości, NAFLD/NASH, cukrzycy typu 2 i raku. Zakres i wpływ „niekanonicznych� ról sygnalizacyjnych ciał ketonowych, w tym regulacja PTM, które prawdopodobnie sprzężą się w przód i w tył na szlaki metaboliczne i sygnalizacyjne, wymagają głębszej eksploracji. Wreszcie, ketogeneza pozawątrobowa może otworzyć intrygujące parakrynne i autokrynne mechanizmy sygnalizacyjne oraz możliwości wpływania na kometabolizm w obrębie układu nerwowego i guzów w celu osiągnięcia celów terapeutycznych.
Podziękowanie
Ncbi.nlm.nih.gov/pmc/articles/PMC5313038/
Przypisy
Podsumowując, ciała ketonowe są tworzone przez wątrobę, aby mogły być wykorzystywane jako źródło energii, gdy w organizmie ludzkim nie ma wystarczającej ilości łatwo dostępnej glukozy. Ketogeneza zachodzi, gdy we krwi występuje niski poziom glukozy, szczególnie po wyczerpaniu innych komórkowych zapasów węglowodanów. Celem powyższego artykułu było omówienie wielowymiarowej roli ciał ketonowych w metabolizmie paliw, sygnalizacji i terapii. Zakres naszych informacji ogranicza się do zagadnień związanych ze zdrowiem kręgosłupa i kręgosłupa. Aby omówić ten temat, prosimy zapytać dr Jimeneza lub skontaktować się z nami pod adresem 915-850-0900 .
Kurator: dr Alex Jimenez
Źródła: Ncbi.nlm.nih.gov/pmc/articles/PMC5313038/

Dodatkowa dyskusja na temat: Ostry ból pleców
Na ból plecówTo jedna z najczęstszych przyczyn niepełnosprawności i nieobecności w pracy na całym świecie. Ból pleców jest drugą najczęstszą przyczyną wizyt lekarskich, ustępującą jedynie infekcjom górnych dróg oddechowych. Około 80 procent populacji przynajmniej raz w życiu doświadczy bólu pleców. Kręgosłup to złożona struktura składająca się z kości, stawów, więzadeł i mięśni oraz innych tkanek miękkich. Urazy i/lub pogorszenie stanu, takie jak�przepukliny, może ostatecznie prowadzić do objawów bólu pleców. Urazy sportowe lub urazy powypadkowe są często najczęstszą przyczyną bólu pleców, jednak czasami najprostsze ruchy mogą mieć bolesne skutki. Na szczęście alternatywne opcje leczenia, takie jak chiropraktyka, mogą pomóc złagodzić ból pleców poprzez zastosowanie korekty kręgosłupa i manipulacji manualnych, ostatecznie poprawiając ulgę w bólu. �

EXTRA EXTRA | WAŻNA TEMAT: Zalecany kręgarz El Paso, TX
***