Back Clinic Wsparcie neurofizjologii klinicznej. El Paso, Teksas. Kręgarz, dr Alexander Jimenez omawia neurofizjologia kliniczna. Dr Jimenez zbada znaczenie kliniczne i czynności funkcjonalne włókien nerwów obwodowych, rdzenia kręgowego, pnia mózgu i mózgu w kontekście zaburzeń trzewnych i mięśniowo-szkieletowych. Pacjenci uzyskają zaawansowaną wiedzę na temat anatomii, genetyki, biochemii i fizjologii bólu w odniesieniu do różnych zespołów klinicznych. Uwzględniona zostanie biochemia żywieniowa związana z nocycepcją i bólem. A wprowadzenie tych informacji do programów terapeutycznych zostanie podkreślone.
Nasz zespół jest dumny z tego, że dostarcza naszym rodzinom i rannym pacjentom tylko sprawdzone protokoły leczenia. Ucząc pełnego, holistycznego dobrego samopoczucia jako stylu życia, zmieniamy nie tylko życie naszych pacjentów, ale także ich rodzin. Robimy to, abyśmy mogli dotrzeć do jak największej liczby mieszkańców El Paso, którzy nas potrzebują, bez względu na problemy z przystępnością. Aby uzyskać odpowiedzi na wszelkie pytania, zadzwoń do dr Jimenez pod numer 915-850-0900.
"Zasady podejmowania decyzji klinicznych, klasyfikacja bólu kręgosłupa i przewidywanie wyników leczenia: omówienie ostatnich doniesień z literatury rehabilitacyjnej"
Abstrakcyjny
Zasady podejmowania decyzji klinicznych są coraz częściej obecne w literaturze biomedycznej i stanowią jedną ze strategii usprawniania podejmowania decyzji klinicznych w celu poprawy wydajności i skuteczności świadczenia opieki zdrowotnej. W kontekście badań nad rehabilitacją, zasady podejmowania decyzji klinicznych miały na celu przede wszystkim klasyfikację pacjentów poprzez przewidywanie ich odpowiedzi na leczenie na określone terapie. Tradycyjnie zalecenia dotyczące opracowywania reguł decyzji klinicznych proponują wieloetapowy proces (wyprowadzenie, walidacja, analiza wpływu) z wykorzystaniem zdefiniowanej metodologii. Wysiłki badawcze mające na celu opracowanie opartej na diagnozie zasady podejmowania decyzji klinicznych odbiegają od tej konwencji. Ostatnie publikacje w tej dziedzinie badań wykorzystywały zmodyfikowaną terminologię, oparty na diagnozie przewodnik podejmowania decyzji klinicznych. Modyfikacje terminologii i metodologii związanej z regułami podejmowania decyzji klinicznych mogą utrudnić klinicystom rozpoznanie poziomu dowodów związanych z regułą podejmowania decyzji i zrozumienie, w jaki sposób te dowody powinny być wdrażane w celu informowania opieki nad pacjentem. Przedstawiamy krótki przegląd rozwoju zasad podejmowania decyzji klinicznych w kontekście literatury dotyczącej rehabilitacji oraz dwóch konkretnych artykułów opublikowanych niedawno w Chiropractic and Manual Therapies.
Zasady przewidywania klinicznego
Opieka zdrowotna przeszła ważną zmianę paradygmatu w kierunku praktyki opartej na dowodach. Podejście, które ma usprawnić podejmowanie decyzji klinicznych poprzez połączenie najlepszych dostępnych dowodów z wiedzą kliniczną i preferencjami pacjentów.
Ostatecznie celem praktyki opartej na dowodach jest poprawa świadczenia opieki zdrowotnej. Jednak przełożenie dowodów naukowych na praktykę okazało się trudnym przedsięwzięciem.
Reguły decyzji klinicznych (CDR), znane również jako reguły przewidywania klinicznego, są coraz częściej spotykane w literaturze dotyczącej rehabilitacji.
Są to narzędzia zaprojektowane w celu informowania o podejmowaniu decyzji klinicznych poprzez identyfikację potencjalnych predyktorów wyniku testu diagnostycznego, rokowania lub odpowiedzi terapeutycznej.
W literaturze dotyczącej rehabilitacji, CDR są najczęściej używane do przewidywania odpowiedzi pacjenta na leczenie. Zaproponowano im identyfikację klinicznie istotnych podgrup pacjentów prezentujących poza tym niejednorodne zaburzenia, takie jak niespecyficzna szyja lub niskie ból pleców, to jest perspektywa, na której zamierzamy się skoncentrować.
Zasady przewidywania klinicznego
Umiejętność klasyfikowania lub podgrupowania pacjentów z niejednorodnymi zaburzeniami, takimi jak ból kręgosłupa, została podkreślona jako priorytet badawczy i, w konsekwencji, przedmiot wielu wysiłków badawczych. Atrakcyjność takich podejść do klasyfikacji polega na ich możliwości poprawienia wydajności i skuteczności leczenia poprzez dopasowanie pacjentów do optymalnych terapii. W przeszłości klasyfikacja pacjentów opierała się na ukrytych podejściach opartych na tradycji lub niesystematycznych obserwacjach. Wykorzystanie CDR do informowania o klasyfikacji jest jedną z prób podejścia bardziej opartego na dowodach, mniej zależnego od nieuzasadnionej teorii.
CDR są opracowywane w wieloetapowym procesie obejmującym badania wyprowadzania, walidacji i analizy wpływu, z których każdy ma określony cel i kryteria metodologiczne. Podobnie jak w przypadku wszystkich form dowodów wykorzystywanych do podejmowania decyzji dotyczących pacjentów, zwrócenie uwagi na odpowiednią metodologię badania ma kluczowe znaczenie dla oceny potencjalnych korzyści z wdrożenia.
Korzyści z zasad przewidywania klinicznego
Może pomieścić więcej czynników niż ludzki mózg może wziąć pod uwagę
Model CDR / CPR zawsze da taki sam wynik (równanie matematyczne)
Może być dokładniejsze niż ocena kliniczna.
Kliniczne zastosowania zasad przewidywania klinicznego
Ostatecznie użyteczność CDR polega nie na jej dokładności, ale na jej zdolności do poprawy wyników klinicznych i zwiększenia skuteczności opieki.[15] Nawet jeśli CDR wykazuje szeroką walidację, nie gwarantuje to, że zmieni on podejmowanie decyzji klinicznych ani że wprowadzone przez nią zmiany spowodują lepszą opiekę.
Zmiany, które powoduje, spowodują lepszą opiekę. McGinn i in. [2] zidentyfikowano trzy wyjaśnienia niepowodzenia CDR na tym etapie. Po pierwsze, jeśli ocena klinicysty jest tak dokładna, jak decyzja oparta na CDR, nie ma korzyści z jej stosowania. Po drugie, zastosowanie CDR może wiązać się z uciążliwymi obliczeniami lub procedurami, które zniechęcają klinicystów do korzystania z CDR. Po trzecie, korzystanie z CDR może nie być wykonalne we wszystkich środowiskach i okolicznościach. Ponadto uwzględnilibyśmy fakt, że badania eksperymentalne mogą obejmować pacjentów, którzy nie są w pełni reprezentatywni dla pacjentów leczonych rutynowo i że może to ograniczać rzeczywistą wartość CDR. Dlatego, aby w pełni zrozumieć użyteczność CDR i jej zdolność do poprawy świadczenia opieki zdrowotnej, konieczne jest przeprowadzenie pragmatycznego zbadania jego wykonalności i wpływu, gdy jest stosowany w środowisku odzwierciedlającym rzeczywistą praktykę. Można to przeprowadzić za pomocą różnych projektów badań, takich jak badania randomizowane, badania randomizowane w klastrach lub inne podejścia, takie jak badanie wpływu CDR przed i po jego wdrożeniu.
Częstość występowania metod klasyfikacji u pacjentów z zaburzeniami lędźwiowymi z wykorzystaniem zespołów McKenziego, schematu bólowego, manipulacji i stabilizacji stanu klinicznego.
Celem było (1) określenie odsetka pacjentów z zaburzeniami lędźwiowymi, którzy mogliby być sklasyfikowani na przyjęciu przez zespoły McKenzie'ego (McK) i klasyfikację bólu (PPC) za pomocą metod oceny diagnostycznej i terapii mechanicznej (MDT), manipulacji i stabilizacji prognoz klinicznych zasady (CPR) i (2) dla każdej kategorii CPR lub kategorii CPR Stab określają częstość występowania klasyfikacji za pomocą McK i PPC.
CPR to wyrafinowane modele probabilistyczne i prognostyczne, w których grupa zidentyfikowanych cech pacjentów i objawów klinicznych jest statystycznie powiązana ze znaczącą prognozą wyników pacjentów.
Badacze opracowali dwie oddzielne resuscytacji krążeniowo-oddechowej w celu identyfikacji pacjentów, którzy reagowaliby korzystnie na manipulacje.33,34 Flynn i in. opracował oryginalną manipulację RKO przy użyciu pięciu kryteriów, tj. brak objawów poniżej kolana, niedawny początek objawów (<16 dni), niski wynik kwestionariusza przekonania o unikaniu strachu36 dla pracy (<19), hipomobilność odcinka lędźwiowego kręgosłupa i wewnętrzna część biodra rotacja ROM (>35 dla co najmniej jednego biodra).33
RKO Flynna została następnie zmodyfikowana przez Fritza i in. dwóm kryteriom, które obejmowały brak objawów poniżej kolana i niedawny początek objawów (<16 dni), jako pragmatyczną alternatywę w celu zmniejszenia obciążenia klinicysty przy identyfikowaniu pacjentów w podstawowej opiece zdrowotnej, którzy z największym prawdopodobieństwem zareagują na manipulację ciągiem [34].
Reguła przewidywania klinicznego (RKO) jest kombinacją wyników klinicznych, które statystycznie wykazały znaczącą przewidywalność w określaniu wybranego stanu lub rokowania pacjenta, który otrzymał określone leczenie 1,2. Resuscytacji krążeniowo-oddechowej tworzy się przy użyciu wielowymiarowych metod statystycznych, ma na celu zbadanie zdolności predykcyjnych wybranych grup zmiennych klinicznych3,4 i ma na celu pomóc klinicystom w podejmowaniu szybkich decyzji, które normalnie mogą podlegać błędnym błędom5. Reguły mają charakter algorytmiczny i obejmują skondensowane informacje, które identyfikują najmniejszą liczbę statystycznie diagnostycznych wskaźników do docelowego stanu6.
Zasady przewidywania klinicznego są na ogół opracowywane przy użyciu metody 3-etapowej14. Po pierwsze, resuscytacja krążeniowo-oddechowa wyprowadziła nas prospektywnie-
wielowymiarowych metod statystycznych w celu zbadania zdolności predykcyjnej wybranych grup zmiennych klinicznych3. Drugi krok obejmuje walidację RKO w randomizowanym, kontrolowanym badaniu w celu zmniejszenia ryzyka, że czynniki prognostyczne rozwinięte w fazie wyprowadzenia zostały wybrane przypadkowo14. Trzeci krok obejmuje przeprowadzenie analizy wpływu, aby określić, w jaki sposób RKO poprawia opiekę, obniża koszty i dokładnie określa docelowy cel14.
Chociaż nie ma dyskusji, że starannie skonstruowane RKO mogą poprawić praktykę kliniczną, według mojej wiedzy, nie ma wytycznych, które określałyby wymagania metodologiczne dla RKO do wlewu we wszystkich środowiskach klinicznych. Wytyczne są tworzone w celu poprawy rygoru projektowania badań i sprawozdawczości. Poniższy artykuł wstępny przedstawia potencjalne pułapki metodologiczne w RKO, które mogą znacząco osłabić możliwość przenoszenia algorytmu. W zakresie rehabilitacji większość RKO ma charakter nakazowy; w związku z tym moje komentarze odzwierciedlają nakazowe CPR.
Pułapki metodologiczne
RKO mają na celu określenie jednorodnego zestawu cech z niejednorodnej populacji prospektywnie wybranych kolejnych pacjentów5,15. Zazwyczaj uzyskana stosowna populacja jest małym podzbiorem większej próby i może stanowić tylko niewielki procent rzeczywistego dziennego obciążenia klinicysty przypadkami. Ustawienie i umiejscowienie większej próby powinno dać się uogólnić15,16, a późniejsze badania trafności wymagają oceny RKO w różnych grupach pacjentów, w różnych środowiskach i z typową grupą pacjentów widzianą przez większość klinicystów16. Ponieważ wiele resuscytacji krążeniowo-oddechowej jest opracowywanych w oparciu o bardzo odmienną grupę, która może, ale nie musi odzwierciedlać typowej populacji pacjentów, zakres możliwości przenoszenia17 wielu obecnych algorytmów resuscytacji krążeniowo-oddechowej może być ograniczony.
Reguły prognozowania klinicznego wykorzystują miary wyników w celu określenia skuteczności interwencji. Miary wyników muszą mieć jedną definicję operacyjną5 i wymagać wystarczającej reakcji, aby naprawdę uchwycić odpowiednią zmianę stanu14; ponadto miary te powinny mieć dobrze skonstruowany punkt odcięcia16,18 i być zbierane przez zaślepionego administratora15. Wybór odpowiedniej punktacji kotwicy do pomiaru rzeczywistych zmian jest obecnie przedmiotem dyskusji19-20. Większość miar wyników wykorzystuje kwestionariusze oparte na przypominaniu pacjentów, takie jak globalna ocena zmian (GRoC), która jest odpowiednia, gdy jest stosowana w krótkim okresie, ale jest obciążona błędem przypominania, gdy jest stosowana w analizach długoterminowych19-21.
Potencjalną wadą resuscytacji krążeniowo-oddechowej jest brak utrzymania jakości testów i miar wykorzystywanych jako predyktory w algorytmie. Dlatego test perspektywiczny i miary powinny być od siebie niezależne podczas modelowania16; każde powinno być wykonane w sensowny, akceptowalny sposób4; klinicyści lub administratorzy danych powinni być ślepi na miary wyników i stan pacjenta22.
Źródła
Potencjalne pułapki reguł prognozowania klinicznego; The Journal of Manual & Manipulative Therapy Volume 16 Number Two [69]
Jeffrey J Hebert i Julie M Fritz; Zasady podejmowania decyzji klinicznych, klasyfikacja bólu kręgosłupa i przewidywanie wyników leczenia: Omówienie ostatnich doniesień w literaturze dotyczącej rehabilitacji
Depresja jest jednym z najczęstszych problemów ze zdrowiem psychicznym w Stanach Zjednoczonych. Aktualne badania sugerują, że depresja jest wynikiem połączenia aspektów genetycznych, biologicznych, ekologicznych i psychologicznych. Depresja jest poważnym zaburzeniem psychiatrycznym na całym świecie, ze znacznym obciążeniem ekonomicznym i psychologicznym dla społeczeństwa. Na szczęście depresję, nawet najcięższe przypadki, można leczyć. Im wcześniej może się rozpocząć leczenie, tym jest ono skuteczniejsze.
W rezultacie jednak istnieje zapotrzebowanie na solidne biomarkery, które pomogą w poprawie diagnozy w celu przyspieszenia procesu odkrywania leku i/lub leku u każdego pacjenta z zaburzeniem. Są to obiektywne, obwodowe wskaźniki fizjologiczne, których obecność można wykorzystać do przewidywania prawdopodobieństwa wystąpienia lub wystąpienia depresji, stratyfikowania według nasilenia lub symptomatologii, wskazywania predykcji i rokowania lub monitorowania odpowiedzi na interwencje terapeutyczne. Celem niniejszego artykułu jest przedstawienie najnowszych spostrzeżeń, obecnych wyzwań i perspektyw na przyszłość dotyczących odkrycia różnych biomarkery na depresję i jak mogą one pomóc w poprawie diagnozy i leczenia.
Biomarkery depresji: najnowsze spostrzeżenia, obecne wyzwania i perspektywy na przyszłość
Abstrakcyjny
Mnóstwo badań wskazało setki domniemanych biomarkerów depresji, ale jeszcze nie wyjaśniło ich roli w chorobie depresyjnej ani nie ustaliło, co jest nienormalne, u których pacjentów i jak informacje biologiczne można wykorzystać do poprawy diagnozy, leczenia i prognozowania. Ten brak postępu wynika częściowo z natury i niejednorodności depresji, w połączeniu z niejednorodnością metodologiczną w literaturze naukowej oraz dużą liczbą potencjalnych biomarkerów, których ekspresja często różni się w zależności od wielu czynników. Dokonujemy przeglądu dostępnej literatury, która wskazuje, że wysoce obiecującymi kandydatami są markery zaangażowane w procesy zapalne, neurotroficzne i metaboliczne, a także komponenty układu neuroprzekaźnikowego i neuroendokrynnego. Można je mierzyć za pomocą ocen genetycznych i epigenetycznych, transkryptomicznych i proteomicznych, metabolomicznych i neuroobrazowych. Obecnie wymagane jest zastosowanie nowatorskich podejść i systematycznych programów badawczych, aby określić, czy i które biomarkery można wykorzystać do przewidywania odpowiedzi na leczenie, podziału pacjentów na określone terapie i opracowywania celów dla nowych interwencji. Dochodzimy do wniosku, że istnieje wiele nadziei na zmniejszenie ciężaru depresji poprzez dalszy rozwój i rozszerzenie tych ścieżek badawczych.
Słowa kluczowe:zaburzenia nastroju, ciężkie zaburzenie depresyjne, stan zapalny, odpowiedź na leczenie, stratyfikacja, medycyna spersonalizowana
Wprowadzenie
Wyzwania w zakresie zdrowia psychicznego i zaburzeń nastroju
Chociaż psychiatria ma większe obciążenie związane z chorobą niż jakakolwiek inna diagnostyczna kategoria medyczna1, nadal widoczna jest dysproporcja w ocenie zdrowia fizycznego i psychicznego w wielu dziedzinach, w tym w zakresie finansowania badań2 i publikacji.3 Wśród trudności, z jakimi boryka się zdrowie psychiczne, jest brak konsensusu wokół klasyfikacji, diagnozy i leczenia, który wynika z niepełnego zrozumienia procesów leżących u podstaw tych zaburzeń. Jest to bardzo widoczne w przypadku zaburzeń nastroju, kategorii, która stanowi największe obciążenie dla zdrowia psychicznego.3 Najbardziej rozpowszechnione zaburzenie nastroju, duże zaburzenie depresyjne (MDD), to złożona, niejednorodna choroba, w której może wystąpić nawet 60% pacjentów. pewien stopień oporności na leczenie, która przedłuża i pogarsza epizody.4 W przypadku zaburzeń nastroju oraz w szerszym zakresie zdrowia psychicznego, wyniki leczenia prawdopodobnie poprawiłyby się dzięki odkryciu solidnych, jednorodnych podtypów w obrębie (i przekrojowych) kategorii diagnostycznych, według których może być rozwarstwiony. Zdając sobie z tego sprawę, obecnie trwają globalne inicjatywy nakreślenia podtypów czynnościowych, takie jak kryteria domeny badawczej.5 Przypuszcza się, że markery biologiczne są priorytetowymi kandydatami do określania podtypów zaburzeń psychicznych.6
Poprawa odpowiedzi na leczenie depresji
Pomimo szerokiego zakresu opcji leczenia dużej depresji, tylko około jedna trzecia pacjentów z MDD osiąga remisję nawet po otrzymaniu optymalnego leczenia przeciwdepresyjnego zgodnie z konsensusowymi wytycznymi i stosując opiekę opartą na pomiarach, a wskaźniki odpowiedzi na leczenie wydają się spadać z każdym nowym leczeniem .7 Co więcej, depresja oporna na leczenie (TRD) wiąże się ze zwiększonym upośledzeniem czynnościowym, śmiertelnością, zachorowalnością oraz nawracającymi lub przewlekłymi epizodami w perspektywie długoterminowej.8,9 Zatem uzyskanie poprawy odpowiedzi na leczenie na dowolnym etapie klinicznym przyniosłoby szersze korzyści dla ogólne wyniki w depresji. Pomimo znacznego obciążenia, jakie przypisuje się TRD, badania w tym obszarze są skąpe. Definicje TRD nie są wystandaryzowane, pomimo wcześniejszych prób4: niektóre kryteria wymagają tylko jednego badania leczenia, w którym nie osiąga się 50% redukcji objawów (z zwalidowanej miary nasilenia depresji), podczas gdy inne wymagają nieosiągnięcia pełnej remisji lub brak odpowiedzi na co najmniej dwa odpowiednio przebadane leki przeciwdepresyjne z różnych klas w ramach epizodu, które należy uznać za TRD.4,10 Ponadto ocena stopnia zaawansowania i przewidywanie oporności na leczenie jest poprawione przez dodanie kluczowych cech klinicznych nasilenia i przewlekłości do liczby nieudanych terapii .9,11 Niemniej jednak ta niespójność definicji sprawia, że interpretacja literatury badawczej dotyczącej TRD staje się jeszcze bardziej złożonym zadaniem.
W celu poprawy odpowiedzi na leczenie, wyraźnie pomocna jest identyfikacja predykcyjnych czynników ryzyka braku odpowiedzi. Scharakteryzowano niektóre ogólne predyktory TRD, w tym brak pełnej remisji po poprzednich epizodach, współwystępujący lęk, samobójstwo i wczesny początek depresji, a także osobowość (szczególnie niska ekstrawersja, niska zależność od nagrody i wysoka neurotyczność) oraz czynniki genetyczne.12 Wyniki te potwierdzają przeglądy, w których zsyntetyzowano oddzielnie dowody dotyczące farmakologicznego13 i psychologicznego14 leczenia depresji. Leki przeciwdepresyjne i terapie poznawczo-behawioralne wykazują w przybliżeniu porównywalną skuteczność,15 ale ze względu na ich różne mechanizmy działania można oczekiwać, że będą miały różne predyktory odpowiedzi. Chociaż trauma we wczesnym okresie życia od dawna wiąże się z gorszymi wynikami klinicznymi i zmniejszoną odpowiedzią na leczenie,16 wczesne wskazania sugerują, że osoby z historią traumy w dzieciństwie mogą lepiej reagować na terapie psychologiczne niż farmakologiczne.17 Mimo to dominuje niepewność i niewielka personalizacja lub stratyfikacja leczenia osiągnęła praktykę kliniczną.18
Niniejszy przegląd skupia się na dowodach potwierdzających użyteczność biomarkerów jako potencjalnie użytecznych narzędzi klinicznych do poprawy odpowiedzi na leczenie depresji.
Biomarkery: systemy i źródła
Biomarkery stanowią potencjalny cel dla identyfikacji predyktorów odpowiedzi na różne interwencje.19 Dotychczasowe dowody sugerują, że markery odzwierciedlające aktywność układów zapalnych, neuroprzekaźników, neurotroficznych, neuroendokrynnych i metabolicznych mogą być w stanie przewidzieć stan zdrowia psychicznego i fizycznego u osób aktualnie cierpiących na depresję , ale istnieje wiele niespójności między odkryciami.20 W tym przeglądzie skupiamy się na tych pięciu układach biologicznych.
Aby w pełni zrozumieć szlaki molekularne i ich udział w zaburzeniach psychicznych, obecnie uważa się, że należy ocenić wiele „poziomów” biologicznych, co jest popularnie określane jako podejście „omiczne”.21 Rycina 1 przedstawia obraz różnych poziomy biologiczne, na których można ocenić każdy z pięciu systemów, oraz potencjalne źródła markerów, na których można przeprowadzić te oceny. Należy jednak pamiętać, że chociaż każdy system można sprawdzić na każdym poziomie omicznym, optymalne źródła pomiaru wyraźnie różnią się na każdym poziomie. Na przykład neuroobrazowanie zapewnia platformę do pośredniej oceny struktury lub funkcji mózgu, podczas gdy badania białek we krwi bezpośrednio oceniają markery. Transkryptomika22 i metabolomika23 cieszą się coraz większą popularnością, oferując ocenę potencjalnie ogromnej liczby markerów, a projekt Human Microbiome Project próbuje obecnie zidentyfikować wszystkie mikroorganizmy i ich skład genetyczny u ludzi.24 Nowatorskie technologie zwiększają naszą zdolność do ich pomiaru, w tym poprzez dodatkowe źródła ; na przykład, hormony, takie jak kortyzol, można teraz oznaczać we włosach lub paznokciach (zapewniając przewlekłą wskazówkę) lub pocie (zapewniając ciągły pomiar),25 jak również we krwi, płynie mózgowo-rdzeniowym, moczu i ślinie.
Biorąc pod uwagę liczbę przypuszczalnych źródeł, poziomów i systemów zaangażowanych w depresję, nie jest zaskakujące, że skala biomarkerów o potencjale translacyjnym jest szeroka. W szczególności, gdy weźmie się pod uwagę interakcje między markerami, jest prawdopodobnie mało prawdopodobne, aby badanie pojedynczych biomarkerów oddzielnie przyniosło wyniki owocne dla poprawy praktyki klinicznej. Schmidt i wsp.26 zaproponowali zastosowanie paneli biomarkerów, a następnie Brand i wsp.27 przedstawili projekt panelu w oparciu o wcześniejsze dowody kliniczne i przedkliniczne dotyczące MDD, identyfikując 16 „silnych” celów biomarkerowych, z których każdy rzadko jest pojedynczym markerem. Obejmują one zmniejszoną objętość istoty szarej (w hipokampie, korze przedczołowej i zwojach podstawy), zmiany cyklu okołodobowego, hiperkortyzolizm i inne objawy nadaktywności osi podwzgórze-przysadka-nadnercza (HPA), dysfunkcję tarczycy, zmniejszoną ilość dopaminy, noradrenaliny lub kwasu 5-hydroksyindolooctowego , podwyższony poziom glutaminianu, podwyższony poziom dysmutazy ponadtlenkowej i peroksydacji lipidów, atenuowany cykliczny 3',5'-monofosforan adenozyny i aktywność szlaku kinazy białkowej aktywowanej mitogenem, podwyższony poziom cytokin prozapalnych, zmiany tryptofanu, kinureniny, insuliny i specyficzne polimorfizmy genetyczne. Te markery nie zostały uzgodnione w drodze konsensusu i mogą być mierzone na różne sposoby; jasne jest, że skoncentrowana i systematyczna praca musi zająć się tym ogromnym zadaniem, aby udowodnić korzyści kliniczne.
Cele tego przeglądu
Jako celowo obszerny przegląd, ten artykuł ma na celu określenie ogólnych potrzeb w zakresie badań nad biomarkerami w depresji oraz zakresu, w jakim biomarkery mają rzeczywisty potencjał translacyjny w zakresie wzmacniania odpowiedzi na leczenie. Zaczynamy od omówienia najważniejszych i najbardziej ekscytujących odkryć w tej dziedzinie i kierujemy czytelnika do bardziej szczegółowych recenzji dotyczących odpowiednich markerów i porównań. Przedstawiamy aktualne wyzwania, przed którymi stoimy w świetle dowodów, w połączeniu z potrzebami zmniejszenia ciężaru depresji. Wreszcie, spoglądamy w przyszłość na ważne ścieżki badawcze umożliwiające sprostanie aktualnym wyzwaniom i ich implikacje dla praktyki klinicznej.
Najnowsze informacje
Poszukiwanie klinicznie użytecznych biomarkerów dla osób z depresją wywołało szeroko zakrojone badania w ciągu ostatniego półwiecza. Najczęściej stosowane terapie wywodziły się z monoaminowej teorii depresji; następnie wiele uwagi zyskały hipotezy neuroendokrynne. W ostatnich latach najbardziej płodne badania dotyczyły zapalnej hipotezy depresji. Jednak duża liczba odpowiednich artykułów przeglądowych dotyczy wszystkich pięciu systemów; patrz Tabela 1 i poniżej, aby zapoznać się z najnowszymi spostrzeżeniami na temat systemów biomarkerów. Chociaż mierzone na wielu poziomach, białka krwiopochodne zostały zbadane najszerzej i stanowią źródło biomarkera, które jest wygodne, opłacalne i może być bliższe potencjałowi translacyjnemu niż inne źródła; w ten sposób bardziej szczegółowo podano biomarkery krążące we krwi.
W niedawnym przeglądzie systematycznym Jani i wsp.20 zbadali biomarkery depresji pochodzące z krwi obwodowej w powiązaniu z wynikami leczenia. Spośród tylko 14 włączonych badań (przeszukiwanych do początku 2013 r.), zbadano 36 biomarkerów, z których 12 było istotnymi predyktorami wskaźników reakcji psychicznej lub fizycznej w co najmniej jednym badaniu. Te zidentyfikowane jako potencjalnie reprezentujące czynniki ryzyka braku odpowiedzi obejmowały białka zapalne: niski poziom interleukiny (IL)-12p70, stosunek liczby limfocytów do liczby monocytów; markery neuroendokrynne (niesupresja kortyzolu przez deksametazon, wysoki poziom krążącego kortyzolu, obniżony poziom hormonu tyreotropowego); markery neuroprzekaźników (niska serotonina i noradrenalina); metaboliczne (cholesterol lipoprotein o niskiej gęstości) i neurotroficzne (obniżone białko B wiążące wapń S100). Ponadto w innych przeglądach opisano związek między dodatkowymi biomarkerami a wynikami leczenia.19,28 Krótki opis przypuszczalnych markerów w każdym systemie przedstawiono w kolejnych rozdziałach oraz w Tabeli 30.
Stany zapalne w depresji
Od czasu przełomowego artykułu Smitha przedstawiającego hipotezę makrofagów,31 ta uznana literatura wskazuje na zwiększone poziomy różnych markerów prozapalnych u pacjentów z depresją, które zostały szeroko omówione.32 W metaanalizach porównujących osoby z depresją i zdrowych oceniono 37 białek zapalnych. populacje kontrolne.38
IL-6 (P<0.001 we wszystkich metaanalizach; 31 badań w tym) i CRP (P<0.001; 20 badań) są często i niezawodnie podwyższone w depresji.40 Podwyższony czynnik martwicy nowotworu alfa (TNF?) został zidentyfikowany we wczesnych badaniach (P<0.001),38, ale znaczna heterogeniczność sprawia, że jest to niejednoznaczne, gdy uwzględnia się nowsze badania (31 badań).40 IL-1? jest jeszcze bardziej niejednoznacznie związany z depresją, a metaanalizy sugerują wyższy poziom w depresji (P=0.03)41, wysoki poziom tylko w badaniach europejskich42 lub brak różnic w porównaniu z grupą kontrolną.40 Mimo to niedawny artykuł sugerował szczególne implikacje translacyjne dla IL- 1?,44 poparte niezwykle istotnym efektem podwyższonego poziomu IL-1? kwas rybonukleinowy przewiduje słabą odpowiedź na leki przeciwdepresyjne;45 inne powyższe wyniki dotyczą krążących cytokin krwiopochodnych. W jednej metaanalizie wykazano zwiększenie stężenia białka chemoatraktantu monocytów chemokiny-1 u uczestników z depresją.39 Interleukiny IL-2, IL-4, IL-8, IL-10 i interferon gamma nie różniły się istotnie między pacjentami z depresją a grupą kontrolną w poziom metaanalityczny, ale mimo to wykazali potencjał pod względem zmian w trakcie leczenia: prospektywnie i przekrojowo zgłaszano podwyższenie IL-8 u osób z ciężką depresją,46 różne wzorce zmian IL-10 i interferonu gamma podczas leczenia IL-47 i IL-4 zmniejszyły się wraz z remisją objawów.2 W metaanalizach wykazano niewielki spadek stężenia IL-48, IL-6β, IL- 1 i CRP.10 Dodatkowo TNF? może zmniejszyć się tylko wraz z leczeniem u osób odpowiadających na leczenie, a złożony wskaźnik markera może wskazywać na nasilenie stanu zapalnego u pacjentów, którzy następnie nie reagują na leczenie.43,49,50 Należy jednak zauważyć, że prawie wszystkie badania dotyczące białek zapalnych i odpowiedzi na leczenie wykorzystują próby leczenia farmakologicznego . Tak więc przynajmniej niektóre zmiany zapalne podczas leczenia można prawdopodobnie przypisać lekom przeciwdepresyjnym. Dokładne działanie przeciwdepresyjne różnych leków przeciwdepresyjnych nie zostało jeszcze ustalone, ale dowody z wykorzystaniem poziomów CRP sugerują, że osoby reagują inaczej na określone terapie w oparciu o stan zapalny. psychoterapia), ale dobra odpowiedź na nortryptylinę lub fluoksetynę; Uher i wsp. 43 powtórzyli to odkrycie dla nortryptyliny i zidentyfikowali odwrotny efekt dla escitalopramu. W przeciwieństwie do tego, Chang i wsp.51 stwierdzili wyższe CRP u osób wcześnie odpowiadających na fluoksetynę lub wenlafaksynę niż u osób nieodpowiadających na leczenie. Co więcej, pacjenci z TRD i wysokim CRP lepiej zareagowali na TNF? antagonistą infliksymabu niż te, których poziom jest w normie.52
Łącznie dowody sugerują, że nawet przy uwzględnieniu czynników takich jak wskaźnik masy ciała (BMI) i wiek, reakcje zapalne wydają się nieprawidłowe u około jednej trzeciej pacjentów z depresją.55,56 Układ zapalny jest jednak niezwykle złożony i istnieje wiele biomarkerów reprezentujących różne aspekty tego systemu. Ostatnio dodatkowe nowe cytokiny i chemokiny dostarczyły dowodów na nieprawidłowości w depresji. Należą do nich: białko hamujące makrofagi 1a, IL-1a, IL-7, IL-12p70, IL-13, IL-15, eotaksyna, czynnik stymulujący tworzenie kolonii makrofagów granulocytów,57 IL-5,58 IL-16,59 IL- 17,60 białko chemoatraktant monocytów-4,61 grasica i chemokina regulowana aktywacją,62 eotaksyna-3, TNFb63 białko indukowane interferonem gamma 10,64 surowiczy amyloid A65 rozpuszczalna wewnątrzkomórkowa cząsteczka adhezyjna66 i rozpuszczalna cząsteczka adhezyjna komórek naczyń 1.67
Ustalenia czynników wzrostu w depresji
W świetle potencjalnego znaczenia nieneurotroficznych czynników wzrostu (takich jak te związane z angiogenezą), odwołujemy się do biomarkerów neurogennych w ramach szerszej definicji czynników wzrostu.
Najczęściej badanym z nich jest czynnik neurotroficzny pochodzenia mózgowego (BDNF). Liczne metaanalizy wykazują osłabienie białka BDNF w surowicy, które wydaje się wzrastać wraz z leczeniem lekami przeciwdepresyjnymi.68 Najnowsza z tych analiz sugeruje, że te aberracje BDNF są bardziej wyraźne u pacjentów z najcięższą depresją, ale wydaje się, że leki przeciwdepresyjne zwiększać poziomy tego białka nawet przy braku klinicznej remisji.71 proBDNF był słabiej badany niż dojrzała forma BDNF, ale wydaje się, że obie te postacie różnią się funkcjonalnie (pod względem ich wpływu na receptory kinazy tyrozynowej B) i niedawne dowody sugerują, że podczas gdy dojrzały BDNF może być zmniejszony w depresji, proBDNF może być nadprodukowany.70 W metaanalizie stwierdzono również, że czynnik wzrostu nerwów oceniany obwodowo w depresji jest niższy niż w grupie kontrolnej, ale może nie być zmieniony przez leczenie przeciwdepresyjne, mimo że jest najbardziej osłabiony u pacjentów z cięższą depresją.72 Podobne wyniki odnotowano w metaanalizie komórek glejowych.czynnik neurotroficzny pochodzenia liniowego.73
Czynnik wzrostu śródbłonka naczyniowego (VEGF) odgrywa rolę w promowaniu angiogenezy i neurogenezy wraz z innymi członkami rodziny VEGF (np. VEGF-C, VEGF-D) i jest obiecujący w przypadku depresji.75 Pomimo niespójnych dowodów, dwie metaanalizy wykazały niedawno wykazali podwyższenie VEGF we krwi pacjentów z depresją w porównaniu z grupą kontrolną (w 16 badaniach; P<0.001).76,77 Jednak niski poziom VEGF wykryto w TRD78, a wyższy poziom przewidywał brak odpowiedzi na leczenie przeciwdepresyjne.79 Nie jest to zrozumiałe. dlaczego poziomy białka VEGF byłyby podwyższone, ale może to częściowo wynikać z aktywności prozapalnej i/lub wzrostu przepuszczalności bariery krew-mózg w stanach depresyjnych, co powoduje zmniejszoną ekspresję w płynie mózgowo-rdzeniowym.80 Związek między VEGF a odpowiedzią na leczenie jest niejasny ; niedawne badanie nie wykazało związku między VEGF, ani BDNF w surowicy z odpowiedzią lub nasileniem depresji, pomimo jej spadku w połączeniu z leczeniem przeciwdepresyjnym.81 Insulinopodobny czynnik wzrostu-1 jest dodatkowym czynnikiem o neurogennych funkcjach, który może być zwiększony w depresji, odzwierciedlając brak równowagi w procesy neurotroficzne.82,83 Podstawowy czynnik wzrostu fibroblastów (lub FGF-2) należy do rodziny czynników wzrostu fibroblastów i wydaje się być wyższy w grupach z depresją niż w grupie kontrolnej.84 Jednak doniesienia nie są spójne; jeden stwierdził, że to białko było niższe w MDD niż zdrowe osoby kontrolne, ale dalej zmniejszało się wraz z leczeniem przeciwdepresyjnym.85
Inne czynniki wzrostu, które nie zostały wystarczająco zbadane w depresji, obejmują kinazę tyrozynową 2 i rozpuszczalną fms-podobną kinazę tyrozynową-1 (zwaną również sVEGFR-1), które działają w synergii z VEGF, a receptory kinazy tyrozynowej (wiążące BDNF) mogą być osłabione w depresji.86 Łożyskowy czynnik wzrostu jest również częścią rodziny VEGF, ale według naszej wiedzy nie był badany w próbkach z systematyczną depresją.
Wyniki badań biomarkerów metabolicznych w depresji
Główne biomarkery związane z chorobami metabolicznymi to leptyna, adiponektyna, grelina, triglicerydy, lipoproteina o dużej gęstości (HDL), glukoza, insulina i albumina.87 Przeanalizowano związki między wieloma z nich a depresją: leptyna88 i grelina89 wydają się słabsze w przypadku depresji. niż w grupie kontrolnej na obrzeżach i może wzrastać wraz z leczeniem przeciwdepresyjnym lub remisją. Insulinooporność może być zwiększona w depresji, aczkolwiek w niewielkich ilościach.90 Profile lipidów, w tym cholesterolu HDL, wydają się być zmienione u wielu pacjentów z depresją, w tym u osób bez współistniejących chorób fizycznych, chociaż związek ten jest złożony i wymaga dalszego wyjaśnienia.91 W przeglądach opisywano hiperglikemię92 i hipoalbuminemię93 w depresji.
Badania ogólnych stanów metabolicznych stają się coraz częstsze przy użyciu paneli metabolomicznych małych cząsteczek w nadziei na znalezienie solidnej sygnatury biochemicznej zaburzeń psychicznych. W niedawnym badaniu z wykorzystaniem modelowania sztucznej inteligencji zestaw metabolitów ilustrujących zwiększoną sygnalizację glukoza-lipid był wysoce predykcyjny dla diagnozy MDD,94 potwierdzając wcześniejsze badania.95
Wyniki neuroprzekaźników w depresji
Chociaż uwaga poświęcona monoaminom w depresji przyniosła stosunkowo skuteczne terapie, nie zidentyfikowano solidnych markerów neuroprzekaźników, które mogłyby zoptymalizować leczenie w oparciu o selektywność monoaminowych celów leków przeciwdepresyjnych. Ostatnie prace wskazują, że receptor serotoniny (5-hydroksytryptaminy) 1A jest potencjalnie ważny zarówno dla diagnozy, jak i prognozowania depresji, w oczekiwaniu na nowe techniki genetyczne i obrazowe.96 Istnieją nowe potencjalne terapie ukierunkowane na 5-hydroksytryptaminę; na przykład, stosując podawanie 5-hydroksytryptofanu o powolnym uwalnianiu.97 Zwiększona transmisja dopaminy oddziałuje z innymi neuroprzekaźnikami w celu poprawy wyników poznawczych, takich jak podejmowanie decyzji i motywacja.98 Podobnie, neuroprzekaźniki glutaminian, noradrenalina, histamina i serotonina mogą oddziaływać i aktywować jako część reakcji na stres związanej z depresją; może to zmniejszyć produkcję 5-hydroksytryptaminy poprzez „powódź”. Niedawny przegląd przedstawia tę teorię i sugeruje, że w TRD można to odwrócić (i przywrócić 5-HT) poprzez multimodalne leczenie ukierunkowane na wiele neuroprzekaźników.99 Co ciekawe, wzrost poziomu serotoniny nie zawsze występuje łącznie z terapeutycznymi korzyściami antydepresyjnymi.100 metabolity neuroprzekaźników, takie jak 3-metoksy-4-hydroksyfenyloglikol noradrenaliny lub kwas homowanilinowy dopaminy, często zwiększały się wraz ze zmniejszeniem depresji za pomocą leczenia przeciwdepresyjnego101,102 lub że niskie poziomy tych metabolitów zapowiadają lepszą odpowiedź na Leczenie SSRI.102,103
Wyniki badań neuroendokrynnych w depresji
Kortyzol jest najczęstszym biomarkerem osi HPA badanym w depresji. Liczne przeglądy skupiały się na różnych ocenach aktywności HPA; ogólnie rzecz biorąc, sugeruje to, że depresja jest związana z hiperkortyzolemią i że reakcja wybudzenia kortyzolu jest często osłabiona.104,105 Potwierdza to niedawny przegląd przewlekłego stężenia kortyzolu mierzonego we włosach, potwierdzający hipotezę o nadaktywności kortyzolu w depresji, ale o hipoaktywności w innych chorobach, takich jak jako zespół lęku napadowego.106 Ponadto szczególnie podwyższony poziom kortyzolu może zapowiadać słabszą odpowiedź na leczenie psychologiczne107 i leki przeciwdepresyjne108. Historycznie najbardziej obiecującym markerem neuroendokrynnym prospektywnej odpowiedzi na leczenie był test supresji deksametazonem, w którym brak supresji kortyzolu po podaniu deksametazonu wiąże się z mniejszym prawdopodobieństwem późniejszej remisji. Jednak zjawisko to nie zostało uznane za wystarczająco silne do zastosowania klinicznego. Pokrewne markery hormonu uwalniającego kortykotropinę i hormon adrenokortykotropiny, jak również wazopresyny, są niekonsekwentnie nadmiernie wytwarzane w depresji, a dehydroepiandrosteron jest atenuowany; stosunek kortyzolu do dehydroepiandrosteronu może być podwyższony jako względnie stabilny marker TRD, utrzymujący się po remisji.109 Dysfunkcje hormonów neuroendokrynnych są od dawna związane z depresją, a niedoczynność tarczycy może również odgrywać rolę w obniżonym nastroju.110 Co więcej, reakcje tarczycy mogą normalizacji dzięki skutecznemu leczeniu depresji.111
W ramach powyższego ważne jest również rozważenie ścieżek sygnałowych w różnych układach, takich jak kinaza 3 syntazy glikogenu, kinaza białkowa aktywowana mitogenami i cykliczny 3',5'-monofosforan adenozyny, zaangażowanych w plastyczność synaptyczną112 i modyfikowanych przez leki przeciwdepresyjne.113 potencjalni kandydaci na biomarkery, które obejmują w szczególności systemy biologiczne, są mierzone za pomocą neuroobrazowania lub genetyki. W odpowiedzi na brak solidnych i znaczących różnic genomicznych między populacjami z depresją i bez depresji114 bardziej użyteczne mogą okazać się nowe podejścia genetyczne, takie jak skala poligeniczna115 lub długość telomerów116,117. Dodatkowymi biomarkerami zyskującymi na popularności są badania cykli okołodobowych czy biomarkery chronobiologiczne z różnych źródeł. Aktygrafia może zapewnić obiektywną ocenę aktywności snu i czuwania oraz odpoczynku za pomocą akcelerometru, a urządzenia aktygraficzne mogą coraz częściej mierzyć dodatkowe czynniki, takie jak ekspozycja na światło. Może to być bardziej przydatne do wykrywania niż powszechnie stosowane subiektywne raporty pacjentów i może dostarczyć nowych predyktorów odpowiedzi na leczenie.118 Pytanie, które biomarkery są najbardziej obiecujące do zastosowania translacyjnego, stanowi wyzwanie, które zostanie rozwinięte poniżej.
Aktualne wyzwania
W przypadku każdego z tych pięciu analizowanych systemów neurobiologicznych dowody są podobne: istnieje wiele biomarkerów, które pod pewnymi względami są powiązane z depresją. Te markery są często ze sobą powiązane w złożony, trudny do modelowania sposób. Dowody są niespójne i jest prawdopodobne, że niektóre z nich są epifenomenami innych czynników, a niektóre są ważne tylko w podgrupie pacjentów. Biomarkery mogą być przydatne na różne sposoby (np. te, które przewidują późniejszą odpowiedź na leczenie, te wskazujące, że określone metody leczenia są bardziej skuteczne lub te, które zmieniają się wraz z interwencjami niezależnie od poprawy klinicznej). Potrzebne są nowatorskie metody, aby zmaksymalizować spójność i kliniczną przydatność ocen biologicznych w populacjach psychiatrycznych.
Zmienność biomarkerów
Zmienność biomarkerów w czasie iw różnych sytuacjach dotyczy bardziej niektórych typów (np. proteomiki) niż innych (genomika). Znormalizowane normy dla wielu nie istnieją lub nie są powszechnie akceptowane. Rzeczywiście, wpływ czynników środowiskowych na markery często zależy od składu genetycznego i innych różnic fizjologicznych między ludźmi, których nie można wszystkich wyjaśnić. To sprawia, że ocena aktywności biomarkerów i identyfikacja nieprawidłowości biologicznych jest trudna do interpretacji. Ze względu na liczbę potencjalnych biomarkerów, wiele z nich nie zostało zmierzonych szeroko lub w pełnym panelu obok innych odpowiednich markerów.
Istnieją doniesienia, że wiele czynników zmienia poziomy białek w różnych układach biologicznych u pacjentów z zaburzeniami afektywnymi. Wraz z czynnikami związanymi z badaniami, takimi jak czas i warunki przechowywania (które mogą powodować rozkład niektórych związków), obejmują one mierzoną porę dnia, pochodzenie etniczne, ćwiczenia,119 dietę (np. aktywność mikrobiomu, zwłaszcza pod warunkiem, że większość badań biomarkerów krwi nie wymagają próbki na czczo),120 palenie i używanie substancji psychoaktywnych,121 a także czynniki zdrowotne (takie jak współistniejące choroby zapalne, sercowo-naczyniowe lub inne choroby fizyczne). Na przykład, chociaż nasilenie stanu zapalnego obserwuje się u osób z depresją, ale skądinąd zdrowych, w porównaniu z grupami bez depresji, osoby z depresją, które również mają współistniejący stan związany z układem odpornościowym, często mają nawet wyższy poziom cytokin niż osoby bez depresji lub choroby. prawdopodobne zaangażowanie w związek między biomarkerami, depresją i odpowiedzią na leczenie przedstawiono poniżej.
Stress. Zarówno reakcje endokrynologiczne, jak i odpornościowe odgrywają dobrze znaną rolę w reagowaniu na stres (fizjologiczny lub psychologiczny), a przejściowy stres w czasie pobierania próbek biologicznych jest rzadko mierzony w badaniach naukowych, pomimo zmienności tego czynnika między osobami, która może być uwydatniona przez prąd objawy depresyjne. Zarówno ostre, jak i przewlekłe stresory psychologiczne działają jak wyzwanie immunologiczne, podkreślając reakcje zapalne w krótkim i dłuższym okresie. osoby dorosłej. brak urazu we wczesnym okresie życia.123,124 Zmiany osi HPA wywołane stresem wydają się być powiązane z funkcjami poznawczymi,125,126, jak również z podtypem depresji lub zmiennością genów związanych z HPA.127 Stres ma również krótko- i długoterminowy wpływ na neurogenezę128 i inne układy nerwowe. [129] Nie jest jasne, w jaki sposób trauma z dzieciństwa wpływa na markery biologiczne u dorosłych z depresją s, ale możliwe jest, że stres we wczesnym okresie życia predysponuje niektóre osoby do trwałych reakcji stresowych w wieku dorosłym, które są wzmacniane psychologicznie i/lub biologicznie.
Funkcjonowanie poznawcze. Dysfunkcje neuropoznawcze występują często u osób z zaburzeniami afektywnymi, nawet w przypadku MDD nie leczonych.133 Deficyty poznawcze kumulują się wraz z opornością na leczenie.134 Neurobiologicznie oś HPA129 i układy neurotroficzne135 prawdopodobnie odgrywają kluczową rolę w tym związku. Neuroprzekaźniki noradrenalina i dopamina są prawdopodobnie ważne dla procesów poznawczych, takich jak uczenie się i pamięć.136 Podwyższone reakcje zapalne powiązano ze spadkiem funkcji poznawczych i prawdopodobnie wpływają na funkcjonowanie poznawcze w epizodach depresyjnych,137 oraz w czasie remisji, poprzez różne mechanizmy.138 Rzeczywiście, Krogh i wsp.139 zasugerowali, że CRP jest bardziej związany z wydajnością poznawczą niż z głównymi objawami depresji.
Wiek, płeć i BMI. Brak lub obecność oraz kierunek różnic biologicznych między mężczyznami i kobietami były szczególnie zmienne w dotychczasowych dowodach. Zmienność hormonów neuroendokrynnych między mężczyznami i kobietami oddziałuje z podatnością na depresję.140 W przeglądzie badań dotyczących stanu zapalnego stwierdzono, że kontrolowanie wieku i płci nie wpływało na różnice w kontroli pacjentów pod względem cytokin zapalnych (chociaż związek między IL-6 a depresją zmniejszał się wraz z wiekiem, co jest zgodne z teoriami, że stan zapalny na ogół nasila się wraz z wiekiem).41,141 Różnice VEGF między pacjentami a grupą kontrolną są większe w badaniach oceniających młodsze próby, podczas gdy płeć, BMI i czynniki kliniczne nie miały wpływu na te porównania na poziomie metaanalizy.77 brak dostosowania do BMI we wcześniejszych badaniach stanu zapalnego i depresji wydaje się zaburzać bardzo istotne różnice pomiędzy tymi grupami.41 Wykazano, że powiększona tkanka tłuszczowa stymuluje produkcję cytokin, a także jest ściśle powiązana z markerami metabolicznymi.142 Ponieważ leki psychotropowe może być związany z wei Przyrost wagi i wyższy BMI, które są związane z opornością na leczenie w depresji, to ważny obszar do zbadania.
Leki. W wielu badaniach biomarkerów dotyczących depresji (zarówno przekrojowych, jak i podłużnych) zebrano próbki wyjściowe od uczestników nieleczonych w celu zmniejszenia heterogeniczności. Jednak wiele z tych ocen jest dokonywanych po okresie wypłukiwania leków, co pozostawia potencjalnie istotny czynnik zakłócający rezydualnych zmian w fizjologii, zaostrzony przez szeroki zakres dostępnych terapii, które mogły mieć różny wpływ na stan zapalny. Niektóre badania wykluczyły stosowanie leków psychotropowych, ale nie innych: w szczególności doustna pigułka antykoncepcyjna jest często dozwolona u uczestniczek badań i nie jest kontrolowana w analizach, co ostatnio wskazywało na zwiększenie poziomu hormonów i cytokin.143,144 Kilka badań wskazuje, że antydepresant leki mają wpływ na odpowiedź zapalną,34,43,49,145 oś HPA147, neuroprzekaźnik108 i aktywność neurotroficzną148. Jednak liczne potencjalne metody leczenia depresji mają odmienne i złożone właściwości farmakologiczne, co sugeruje, że różne opcje leczenia mogą mieć dyskretne skutki biologiczne, poparte aktualnymi danymi. Istnieją teorie, że oprócz działania monoaminowego, specyficzne leki ukierunkowane na serotoninę (tj. SSRI) mogą prawdopodobnie działać na przesunięcia Th149 w zapaleniu, a antydepresanty noradrenergiczne (np. SNRI) wpływają na przesunięcie Th2 Nie jest jeszcze możliwe określić wpływ pojedynczych lub złożonych leków na biomarkery. Są one prawdopodobnie za pośrednictwem innych czynników, w tym czasu trwania leczenia (niewiele badań ocenia długoterminowe stosowanie leków), niejednorodności próby i braku stratyfikacji uczestników według odpowiedzi na leczenie.
Niejednorodność
Metodologiczne. Jak wspomniano powyżej, różnice (pomiędzy badaniami i w obrębie badań) pod względem tego, jakie terapie (i kombinacje) uczestnicy biorą i podjęli wcześniej, z pewnością wprowadzą niejednorodność do wyników badań, szczególnie w badaniach biomarkerów. Oprócz tego wiele innych cech projektu i próby różni się w poszczególnych badaniach, zwiększając w ten sposób trudności w interpretacji i przypisywaniu wyników. Obejmują one parametry pomiaru biomarkerów (np. zestawy testowe) oraz metody zbierania, przechowywania, przetwarzania i analizowania markerów w depresji. Hiles i wsp.141 przeanalizowali niektóre źródła niespójności w piśmiennictwie na temat stanu zapalnego i stwierdzili, że dokładność diagnozy depresji, BMI i choroby współistniejące są najważniejsze w ocenie zapalenia obwodowego między grupami z depresją i bez depresji.
Kliniczny. Rozległa heterogeniczność populacji z depresją jest dobrze udokumentowana151 i ma decydujący wpływ na sprzeczne wyniki w literaturze naukowej. Jest prawdopodobne, że nawet w obrębie diagnoz, nieprawidłowe profile biologiczne ograniczają się do podgrup osobników, które mogą nie być stabilne w czasie. Spójne podgrupy osób cierpiących na depresję można zidentyfikować na podstawie kombinacji czynników psychologicznych i biologicznych. Poniżej przedstawiamy możliwości eksploracji podgrup w sprostaniu wyzwaniom, jakie niosą ze sobą zmienność i heterogeniczność biomarkerów.
Podtypy w depresji
Jak dotąd żadna jednorodna podgrupa w obrębie epizodów lub zaburzeń depresji nie była w stanie wiarygodnie rozróżnić pacjentów na podstawie objawów lub reakcji na leczenie.152 Istnienie podgrupy, w której aberracje biologiczne są bardziej wyraźne, pomogłoby wyjaśnić niejednorodność między poprzednimi badaniami a może katalizować drogę do leczenia warstwowego. Kunugi i wsp.153 zaproponowali zestaw czterech potencjalnych podtypów w oparciu o rolę różnych układów neurobiologicznych wykazujących klinicznie istotne podtypy w depresji: osoby z hiperkortyzolizmem objawiające się depresją melancholijną lub hipokortyzolizm odzwierciedlający podtyp atypowy, podgrupę pacjentów związanych z dopaminą, którzy mogą występują wyraźnie z anhedonią (i mogą dobrze reagować np. na arypiprazol) i podtypem zapalnym charakteryzującym się podwyższonym stanem zapalnym. Wiele artykułów skupiających się na zapaleniu określiło przypadek istnienia „podtypu zapalnego” w depresji.55,56,154,155 Kliniczne korelacje podwyższonego stanu zapalnego nie zostały jeszcze ustalone i podjęto niewiele bezpośrednich prób, aby odkryć, którzy uczestnicy mogą należeć do tej kohorty. Sugerowano, że osoby z depresją atypową mogą mieć wyższy poziom stanu zapalnego niż podtyp melancholijny,156, co być może nie jest zgodne z ustaleniami dotyczącymi osi HPA w podtypach depresji melancholijnej i atypowej. TRD37 lub depresja z wyraźnymi objawami somatycznymi157 również została uznana za potencjalny podtyp zapalny, ale neurowegetatywny (sen, apetyt, utrata libido), nastrój (w tym niski nastrój, myśli samobójcze i drażliwość) oraz objawy poznawcze (w tym uprzedzenia afektywne i poczucie winy)158. wydają się być powiązane z profilami biologicznymi. Kolejni potencjalni kandydaci do podtypu zapalnego obejmują doświadczanie objawów chorobowych podobnych do zachowań159,160 lub zespołu metabolicznego.158
Skłonność do (hipo)manii może biologicznie różnicować pacjentów cierpiących na depresję. Obecnie dostępne dowody wskazują, że choroby afektywne dwubiegunowe są wieloaspektową grupą zaburzeń nastroju, przy czym subsyndromiczna choroba afektywna dwubiegunowa występuje częściej niż wcześniej sądzono.161 Niedokładne i/lub opóźnione wykrywanie choroby afektywnej dwubiegunowej zostało ostatnio podkreślone jako główny problem w psychiatrii klinicznej. średni czas na postawienie diagnozy często przekracza dekadę162, a to opóźnienie powoduje większe nasilenie i koszty całej choroby.163 Ponieważ większość pacjentów z ChAD ma początkowo jeden lub więcej epizodów depresyjnych, a depresja jednobiegunowa jest najczęstszą błędną diagnozą. czynniki, które mogą różnicować depresję jednobiegunową i dwubiegunową, mają istotne implikacje.164 Zaburzenia ze spektrum dwubiegunowego prawdopodobnie nie zostały wykryte w niektórych wcześniejszych badaniach biomarkerów MDD, a nieliczne dowody wskazują na zróżnicowanie aktywności osi HPA109 lub zapalenia165,166 między depresją dwubiegunową i jednobiegunową. sesję. Jednak porównania te są rzadkie, mają małe rozmiary próbek, zidentyfikowano nieistotne efekty trendów lub zrekrutowane populacje, które nie zostały dobrze scharakteryzowane przez diagnozę. Badania te nie analizują również roli odpowiedzi na leczenie w tych związkach.
Zarówno zaburzenia afektywne dwubiegunowe167, jak i oporność na leczenie168 nie są konstruktami dychotomicznymi i leżą na ciągłości, co zwiększa wyzwanie w identyfikacji podtypów. Oprócz podtypowania warto zauważyć, że wiele nieprawidłowości biologicznych obserwowanych w depresji jest podobnie stwierdzanych u pacjentów z innymi rozpoznaniami. Potencjalnie ważne są więc również badania transdiagnostyczne.
Wyzwania związane z pomiarem biomarkerów
Wybór biomarkerów. Duża liczba potencjalnie użytecznych biomarkerów stanowi wyzwanie dla psychobiologii w określeniu, które markery są zaangażowane w jaki sposób i dla kogo. Aby zwiększyć wyzwanie, stosunkowo niewiele z tych biomarkerów poddano wystarczającym badaniom w depresji, a dla większości ich dokładna rola w zdrowych i klinicznych populacjach nie jest dobrze poznana. Mimo to podjęto szereg prób zaproponowania obiecujących paneli biomarkerów. Oprócz Branda i wsp. 16 zestawów markerów o dużym potencjale, 27 Lopresti i wsp. przedstawili dodatkowy obszerny zestaw markerów stresu oksydacyjnego, które mogą poprawić odpowiedź na leczenie.28 Papakostas i wsp. zdefiniowali a priori zestaw dziewięciu markerów surowicy obejmujących systemy biologiczne (BDNF, kortyzol, rozpuszczalny receptor TNFa typu II, alfa1 antytrypsyna, apolipoproteina CIII, naskórkowy czynnik wzrostu, mieloperoksydaza, prolaktyna i rezystyna) w próbkach walidacyjnych i replikacyjnych z MDD. Po połączeniu, złożona miara tych poziomów była w stanie odróżnić grupy MDD od grup kontrolnych z dokładnością 80%–90%.169 Proponujemy, aby nawet one nie obejmowały wszystkich potencjalnych kandydatów w tej dziedzinie; patrz Tabela 2, aby zapoznać się z niewyczerpującym opisem biomarkerów mogących powodować depresję, zawierającym zarówno te posiadające bazę dowodową, jak i obiecujące nowe markery.
Technologia. Dzięki postępowi technologicznemu obecnie możliwe jest (w istocie wygodne) mierzenie dużej liczby biomarkerów jednocześnie przy niższych kosztach iz wyższą czułością niż miało to miejsce wcześniej. Obecnie ta zdolność do pomiaru wielu związków wyprzedza naszą zdolność do efektywnej analizy i interpretacji danych170, co będzie kontynuowane wraz ze wzrostem liczby macierzy biomarkerów i nowych markerów, takich jak metabolomika. Wynika to w dużej mierze z braku zrozumienia dokładnych ról i wzajemnych powiązań między markerami oraz niewystarczającego zrozumienia, jak spokrewnione markery łączą się na różnych poziomach biologicznych (np. genetycznych, transkrypcyjnych, białkowych) w obrębie osób i między nimi. Duże zbiory danych przy użyciu nowych podejść analitycznych i standardów pomogą w rozwiązaniu tego problemu i proponowane są nowe metodologie; jednym z przykładów jest opracowanie podejścia statystycznego opartego na analizie opartej na strumieniu w celu odkrycia nowych potencjalnych markerów metabolicznych w oparciu o ich reakcje między sieciami i zintegrowanie ekspresji genów z danymi dotyczącymi metabolitów.171 Techniki uczenia maszynowego są już stosowane i pomogą w modelach wykorzystujących biomarkery dane do przewidywania wyników leczenia w badaniach z dużymi danymi.172
Agregacja biomarkerów. Jednoczesne badanie szeregu biomarkerów jest alternatywą dla badania izolowanych markerów, która może zapewnić dokładniejszy punkt widzenia na złożoną sieć biologicznych systemów lub sieci.26 Ponadto, aby pomóc w rozplątaniu kontrastujących ze sobą dowodów w tej dotychczasowej literaturze (szczególnie w przypadku sieci biomarkerów a interakcje są dobrze zrozumiane), dane biomarkerów można następnie agregować lub indeksować. Jednym z wyzwań jest zidentyfikowanie optymalnej metody przeprowadzenia tego i może to wymagać ulepszenia technologii i/lub nowatorskich technik analitycznych (patrz sekcja „Big data”). Historycznie, proporcje między dwoma różnymi biomarkerami przyniosły interesujące odkrycia.109,173 Podjęto niewiele prób agregacji danych biomarkerów na większą skalę, na przykład przy użyciu analizy głównych składowych sieci cytokin prozapalnych.174 W metaanalizie wykazano, że cytokiny prozapalne przekształcone w punktację wielkości pojedynczego efektu dla każdego badania i ogólnie wykazały znacznie wyższy stan zapalny przed leczeniem przeciwdepresyjnym, przewidując późniejszy brak odpowiedzi w badaniach ambulatoryjnych. Złożone panele biomarkerów są zarówno wyzwaniem, jak i szansą dla przyszłych badań w celu zidentyfikowania znaczących i wiarygodnych wyników, które można zastosować do poprawy wyników leczenia.43 Badanie przeprowadzone przez Papakostas i wsp. przyjęło alternatywne podejście, wybierając panel heterogenicznych biomarkerów surowicy (zapalnych, Oś HPA i układy metaboliczne), co do których wskazano, że różnią się między osobami z depresją i grupą kontrolną w poprzednim badaniu, i połączono je w wynik ryzyka, który różnił się w dwóch niezależnych próbach i grupie kontrolnej z >80% czułością i swoistością.169
Duże dane. Wykorzystanie dużych zbiorów danych jest prawdopodobnie niezbędne do sprostania obecnym wyzwaniom nakreślonym wokół heterogeniczności, zmienności biomarkerów, identyfikacji optymalnych markerów i skierowania pola w kierunku translacyjnych, stosowanych badań nad depresją. Jednak, jak wskazano powyżej, wiąże się to z wyzwaniami technologicznymi i naukowymi.175 Nauki o zdrowiu dopiero niedawno zaczęły wykorzystywać analitykę dużych zbiorów danych, mniej więcej dekadę później niż w sektorze biznesowym. Jednak badania, takie jak iSPOT-D152 i konsorcja, takie jak Psychiatric Genetics Consortium176, rozwijają się wraz z naszym zrozumieniem mechanizmów biologicznych w psychiatrii. Algorytmy uczenia maszynowego w bardzo niewielu badaniach zaczęto stosować do biomarkerów depresji: w niedawnym badaniu zebrano dane od >5,000 uczestników 250 biomarkerów; po wielokrotnej imputacji danych przeprowadzono regresję wspomaganą przez uczenie maszynowe, wskazując 21 potencjalnych biomarkerów. W wyniku dalszych analiz regresji wybrano trzy biomarkery, które najsilniej wiążą się z objawami depresji (wysoce zmienna wielkość krwinek czerwonych, poziom glukozy i bilirubiny w surowicy). Autorzy wnioskują, że duże zbiory danych mogą być skutecznie wykorzystywane do generowania hipotez.177 Obecnie trwają większe projekty dotyczące fenotypowania biomarkerów, które pomogą nam posunąć naprzód naszą podróż w przyszłość neurobiologii depresji.
Perspektywy na przyszłość
Identyfikacja panelu biomarkerów
Dotychczasowe ustalenia w literaturze wymagają powtórzenia w badaniach na dużą skalę. Jest to szczególnie prawdziwe w przypadku nowych biomarkerów, takich jak chemokina grasica i chemokina regulowana aktywacją oraz kinaza tyrozynowa czynnika wzrostu 2, które, według naszej wiedzy, nie były badane w próbkach kontrolnych ze stanem klinicznym i zdrowych. Badania dużych zbiorów danych muszą analizować kompleksowe panele biomarkerów i wykorzystywać zaawansowane techniki analityczne, aby w pełni ustalić powiązania między markerami a czynnikami, które je modyfikują w populacjach klinicznych i nieklinicznych. Dodatkowo, replikacje analizy głównych składowych na dużą skalę mogą ustalić silnie skorelowane grupy biomarkerów, a także mogą pomóc w stosowaniu „kompozytów” w psychiatrii biologicznej, co może zwiększyć jednorodność przyszłych wyników.
Odkrycie jednorodnych podtypów
Jeśli chodzi o wybór biomarkerów, wiele paneli może być wymaganych dla różnych potencjalnych ścieżek, które mogą implikować badania. Podsumowując, obecne dowody wskazują, że profile biomarkerów są z pewnością, ale w subpopulacji osób cierpiących obecnie na depresję, całkowicie zmienione. Można to ustalić w obrębie kategorii diagnostycznych lub między nimi, co wyjaśniałoby pewną niespójność wyników, którą można zaobserwować w tej literaturze. Ilościowe określenie podgrupy biologicznej (lub podgrup) może być najskuteczniej ułatwione dzięki dużej analizie skupień paneli sieci biomarkerów w depresji. To zilustrowałoby zmienność wewnątrz populacji; Analizy klas utajonych mogą wykazywać wyraźne cechy kliniczne oparte na przykład na zapaleniu.
Specyficzny wpływ leczenia na stan zapalny i odpowiedź
Wszystkie powszechnie przepisywane leki na depresję należy kompleksowo ocenić pod kątem ich specyficznych skutków biologicznych, uwzględniając również skuteczność prób leczenia. Może to umożliwić konstruktom związanym z biomarkerami i prezentacjami objawów przewidywanie wyników różnych terapii przeciwdepresyjnych w bardziej spersonalizowany sposób, co może być możliwe zarówno w kontekście depresji jednobiegunowej, jak i dwubiegunowej. Prawdopodobnie będzie to przydatne w przypadku nowych potencjalnych terapii, a także obecnie wskazanych terapii.
Prospektywne określenie odpowiedzi na leczenie
Zastosowanie powyższych technik prawdopodobnie spowoduje poprawę zdolności do prognozowania oporności na leczenie w sposób prospektywny. Mogą się do tego przyczynić bardziej autentyczne i trwałe (np. długoterminowe) pomiary odpowiedzi na leczenie. Ocena innych ważnych miar dobrostanu pacjenta (takich jak jakość życia i codzienne funkcjonowanie) może zapewnić bardziej holistyczną ocenę wyników leczenia, która może być ściślej powiązana z biomarkerami. Chociaż sama aktywność biologiczna może nie być w stanie odróżnić osób odpowiadających na leczenie od osób nieodpowiadających na leczenie, jednoczesne pomiary biomarkerów ze zmiennymi psychospołecznymi lub demograficznymi można zintegrować z informacjami o biomarkerach w celu opracowania modelu predykcyjnego niewystarczającej odpowiedzi na leczenie. Jeśli zostanie opracowany wiarygodny model do przewidywania odpowiedzi (albo dla populacji z depresją, albo subpopulacji) i zostanie on zweryfikowany retrospektywnie, projekt translacyjny może ustalić jego przydatność w dużym badaniu kontrolowanym.
W kierunku leczenia warstwowego
Obecnie pacjenci z depresją nie są systematycznie kierowani na zoptymalizowany program interwencyjny. W przypadku walidacji, stratyfikowany projekt badania może zostać wykorzystany do przetestowania modelu w celu przewidzenia braku odpowiedzi i/lub określenia, gdzie pacjent musi zostać poddany selekcji w modelu opieki etapowej. Może to być przydatne zarówno w standardowych, jak i naturalistycznych warunkach leczenia, w różnych rodzajach interwencji. Ostatecznie można by opracować klinicznie wykonalny model, aby zapewnić osobom najbardziej odpowiednie leczenie, rozpoznać osoby, u których istnieje prawdopodobieństwo wystąpienia depresji opornej na leczenie, oraz zapewnić lepszą opiekę i monitorowanie tym pacjentom. Pacjentom zidentyfikowanym jako zagrożeni opornością na leczenie można zalecić równoczesną terapię psychologiczną i farmakologiczną lub farmakoterapię skojarzoną. Jako przykład spekulacyjny, uczestnicy bez podwyższenia poziomu cytokin prozapalnych mogą być wskazani do otrzymywania terapii psychologicznej, a nie farmakologicznej, podczas gdy podgrupa pacjentów ze szczególnie wysokim stanem zapalnym może otrzymać środek przeciwzapalny w uzupełnieniu do standardowego leczenia. Podobnie jak w przypadku stratyfikacji, w przyszłości mogą być możliwe spersonalizowane strategie doboru leczenia. Na przykład dana osoba z depresją może mieć znacznie wysoki TNF? poziomy, ale żadnych innych nieprawidłowości biologicznych, i czy może odnieść korzyść z krótkotrwałego leczenia TNF? antagonistów.54 Spersonalizowane leczenie może również obejmować monitorowanie ekspresji biomarkerów podczas leczenia w celu poinformowania o możliwych zmianach w interwencji, wymaganej długości kontynuacji terapii lub w celu wykrycia wczesnych markerów nawrotu.
Nowe cele leczenia
Istnieje ogromna liczba potencjalnych sposobów leczenia depresji, które nie zostały odpowiednio zbadane, w tym nowe lub zmienione interwencje z innych dziedzin medycyny. Niektóre z najpopularniejszych celów znajdowały się w lekach przeciwzapalnych, takich jak celekoksyb (i inne inhibitory cyklooksygenazy-2), TNF? antagoniści etanercept i infliksymab, minocyklina lub aspiryna. Wydaje się to obiecujące.178 Związki antyglukokortykoidowe, w tym ketokonazol179 i metyrapon,180 były badane pod kątem depresji, ale oba mają wady związane z profilem działań niepożądanych, a kliniczny potencjał metyraponu jest niepewny. Mifepriston181 oraz kortykosteroidy fludrokortyzon i spironolakton,182 oraz deksametazon i hydrokortyzon183 mogą również być skuteczne w krótkoterminowym leczeniu depresji. Ukierunkowanie na antagonistów receptora glutaminianu N-metylo-d-asparaginianu, w tym ketaminę, może stanowić skuteczną terapię w depresji.184 Wielonienasycone kwasy tłuszczowe omega-3 wpływają na aktywność zapalną i metaboliczną i wydają się wykazywać pewną skuteczność w leczeniu depresji.185 Możliwe, że statyny mogą mają działanie przeciwdepresyjne186 poprzez odpowiednie szlaki neurobiologiczne.187
W ten sposób biochemiczne działanie leków przeciwdepresyjnych (patrz rozdział „Leki”) zostało wykorzystane do uzyskania korzyści klinicznych w innych dyscyplinach: szczególnie w chorobach gastroenterologicznych, neurologicznych i niespecyficznych objawach.188 Działanie przeciwzapalne leków przeciwdepresyjnych może stanowić część mechanizmu te korzyści. Sugeruje się również, że lit zmniejsza stan zapalny, przede wszystkim poprzez szlaki kinazy 3 syntazy glikogenu.189 Skupienie się na tych efektach może okazać się przydatne dla sygnatury biomarkera depresji, a biomarkery mogą z kolei stanowić markery zastępcze do opracowywania nowych leków.
Wgląd doktora Alexa Jimeneza
Depresja jest zaburzeniem zdrowia psychicznego charakteryzującym się poważnymi objawami wpływającymi na nastrój, w tym utratą zainteresowania aktywnością. Jednak ostatnie badania naukowe wykazały, że możliwe jest zdiagnozowanie depresji na podstawie czegoś więcej niż tylko objawów behawioralnych pacjenta. Zdaniem naukowców identyfikacja łatwo dostępnych biomarkerów, które mogłyby dokładniej diagnozować depresję, ma fundamentalne znaczenie dla poprawy ogólnego stanu zdrowia i dobrego samopoczucia pacjenta. Na przykład, odkrycia kliniczne sugerują, że osoby z poważnym zaburzeniem depresyjnym lub MDD mają niższy poziom cząsteczki acetylo-L-karnityny lub LAC we krwi niż zdrowi kontrolni. Ostatecznie ustalenie biomarkerów depresji może potencjalnie pomóc w lepszym określeniu, kto jest zagrożony rozwojem choroby, a także pomóc pracownikom służby zdrowia w określeniu najlepszej opcji leczenia dla pacjenta z depresją.
Wnioski
Literatura wskazuje, że około dwie trzecie pacjentów z depresją nie osiąga remisji po początkowym leczeniu, a prawdopodobieństwo braku odpowiedzi wzrasta wraz z liczbą badanych terapii. Zapewnienie nieskutecznych terapii ma poważne konsekwencje dla kosztów indywidualnych i społecznych, w tym uporczywego cierpienia i złego samopoczucia, ryzyka samobójstwa, utraty produktywności i marnowania zasobów opieki zdrowotnej. Obszerna literatura dotycząca depresji wskazuje na ogromną liczbę biomarkerów, które mogą usprawnić leczenie osób z depresją. Oprócz neuroprzekaźników i markerów neuroendokrynnych, które od wielu dziesięcioleci są przedmiotem szeroko zakrojonych badań, ostatnie spostrzeżenia wskazują, że odpowiedź zapalna (i ogólniej układ odpornościowy), czynniki metaboliczne i wzrostowe są ważnymi czynnikami zaangażowanymi w depresję. Jednak nadmiernie kontrastujące dowody pokazują, że istnieje szereg wyzwań, które należy rozwiązać, zanim badania nad biomarkerami będą mogły zostać zastosowane w celu poprawy zarządzania i opieki nad osobami z depresją. Ze względu na samą złożoność systemów biologicznych równoczesne badania szerokiego zakresu markerów w dużych próbkach przynoszą znaczną korzyść w odkrywaniu interakcji między stanami biologicznymi i psychologicznymi u poszczególnych osób. Optymalizacja pomiaru zarówno parametrów neurobiologicznych, jak i klinicznych miar depresji prawdopodobnie ułatwi lepsze zrozumienie. W niniejszym przeglądzie podkreślono również znaczenie zbadania czynników potencjalnie modyfikujących (takich jak choroba, wiek, funkcje poznawcze i leki) dla uzyskania spójnego zrozumienia biologii depresji i mechanizmów oporności na leczenie. Jest prawdopodobne, że niektóre markery będą najbardziej obiecujące w przewidywaniu odpowiedzi na leczenie lub oporności na określone leczenie w podgrupie pacjentów, a równoczesny pomiar danych biologicznych i psychologicznych może zwiększyć zdolność do prospektywnej identyfikacji osób zagrożonych słabymi wynikami leczenia. Stworzenie panelu biomarkerów ma wpływ na zwiększenie dokładności diagnostycznej i rokowania, a także indywidualizację leczenia na najwcześniejszym możliwym do zastosowania etapie choroby depresyjnej oraz opracowanie skutecznych nowych celów leczenia. Te implikacje mogą ograniczać się do podgrup pacjentów z depresją. Ścieżki prowadzące do tych możliwości uzupełniają ostatnie strategie badawcze, aby ściślej powiązać zespoły kliniczne z podstawowymi substratami neurobiologicznymi.6 Oprócz zmniejszenia heterogeniczności może to ułatwić przesunięcie w kierunku równorzędności między zdrowiem fizycznym i psychicznym. Oczywiste jest, że chociaż potrzeba dużo pracy, ustalenie związku między odpowiednimi biomarkerami a zaburzeniami depresyjnymi ma istotne implikacje dla zmniejszenia obciążenia depresją na poziomie indywidualnym i społecznym.
Podziękowanie
Niniejszy raport przedstawia niezależne badania finansowane przez Centrum Badań Biomedycznych Narodowego Instytutu Badań nad Zdrowiem (NIHR) w południowym Londynie oraz Maudsley NHS Foundation Trust i King's College London. Wyrażone poglądy są poglądami autorów i niekoniecznie są poglądami NHS, NIHR lub Departamentu Zdrowia.
Przypisy
Ujawnienie. AHY w ciągu ostatnich 3 lat otrzymała honoraria za przemówienie od Astry Zeneca (AZ), Lundbeck, Eli Lilly, Sunovion; honoraria za konsultacje od Allergan, Livanova i Lundbeck, Sunovion, Janssen; oraz wsparcie grantów badawczych od agencji finansujących Janssen i UK (NIHR, MRC, Wellcome Trust). AJC w ciągu ostatnich 3 lat otrzymał honoraria za przemówienie od Astra Zeneca (AZ), honoraria za konsultacje od Allergan, Livanova i Lundbeck oraz wsparcie grantów badawczych od Lundbeck i brytyjskich agencji finansujących (NIHR, MRC, Wellcome Trust).
Autorzy nie zgłaszają innych konfliktów interesów w tej pracy.
PodsumowującChociaż liczne badania naukowe wykazały setki biomarkerów depresji, niewiele z nich ustaliło ich rolę w chorobach depresyjnych lub jak dokładnie można wykorzystać informacje biologiczne do poprawy diagnozy, leczenia i prognozowania. Jednak powyższy artykuł zawiera przegląd dostępnej literatury na temat biomarkerów zaangażowanych w inne procesy i porównuje wyniki kliniczne z objawami depresji. Co więcej, nowe odkrycia dotyczące biomarkerów depresji mogą pomóc w lepszym diagnozowaniu depresji w celu kontynuowania lepszego leczenia. Informacje podane w National Center for Biotechnology Information (NCBI). Zakres naszych informacji ogranicza się do chiropraktyki oraz urazów i schorzeń kręgosłupa. Aby omówić ten temat, prosimy zapytać dr Jimeneza lub skontaktować się z nami pod adresem�915-850-0900 .
Kurator: dr Alex Jimenez
Dodatkowe tematy: Back Pain
Na ból pleców jest jedną z najczęstszych przyczyn niepełnosprawności i nieobecności w pracy na całym świecie. W rzeczywistości, ból pleców został przypisany jako drugi najczęstszy powód wizyt u lekarza, przewyższający jedynie infekcje górnych dróg oddechowych. Około 80 procent populacji doświadczy pewnego rodzaju bólu pleców przynajmniej raz w ciągu całego życia. Kręgosłup jest złożoną strukturą zbudowaną z kości, stawów, więzadeł i mięśni oraz innych miękkich tkanek. Z tego powodu urazy i / lub pogorszenie warunków, takich jak przepukliny, może ostatecznie prowadzić do objawów bólu pleców. Urazy sportowe lub wypadki samochodowe są często najczęstszą przyczyną bólu pleców, jednak czasami najprostsze ruchy mogą mieć bolesne skutki. Na szczęście alternatywne opcje leczenia, takie jak opieka chiropraktyczna, mogą pomóc złagodzić ból pleców poprzez zastosowanie korekcji kręgosłupa i manualnych manipulacji, ostatecznie poprawiając ulgę w bólu.
1.�Prince M, Patel V, Saxena S, et al. Nie ma zdrowia bez zdrowia psychicznego.�Lancet.�2007;370(9590):859.[PubMed]
2.�Kingdon D, Wykes T. Zwiększone fundusze potrzebne na badania nad zdrowiem psychicznym.�BMJ.�2013;346:f402.[PubMed]
3.�Vivekanantham S, Strawbridge R, Rampuri R, Ragunathan T, Young AH. Równość publikacji dla psychiatrii.�Br J Psychiatria.�2016;209(3):257.�[PubMed]
4.�Fava M. Diagnoza i definicja depresji lekoopornej.�Biol Psychiatria.�2003;53(8):649.�[PubMed]
5.�Insel T, Cuthbert B, Garvey M, et al. Kryteria domeny badawczej (RDoC): w kierunku nowych ram klasyfikacji badań nad zaburzeniami psychicznymi.�Jestem J Psychiatrią.�2010;167(7):748.�[PubMed]
6.�Kapur S, Phillips AG, Insel TR. Dlaczego opracowanie testów klinicznych zajęło psychiatrii biologicznej tak długo i co z tym zrobić?Mol Psychiatria.�2012;17(12):1174.�[PubMed]
7.�Gaynes BN, Strażnik D, Trivedi MH, Wiśniewski SR, Fava M, Rush JA. Czego nauczył nas STAR*D? Wyniki dużego, praktycznego badania klinicznego u pacjentów z depresją.�Psychiatr Serw.�2009;60(11):1439.�[PubMed]
8.�Fekadu A, Rane LJ, Wooderson SC, Markopoulou K, Poon L, Cleare AJ. Przewidywanie długoterminowego wyniku lekoopornej depresji w opiece wyższej.�Br J Psychiatria.�2012;201(5):369.[PubMed]
9.�Fekadu A, Wooderson SC, Markopoulo K, Donaldson C, Papadopoulos A, Cleare AJ. Co dzieje się z pacjentami z depresją lekooporną? Systematyczny przegląd średnio- i długoterminowych badań wyników.�J Zaburzenia afektu.�2009;116(1):2.�[PubMed]
10.�Trivedi M. Strategie leczenia mające na celu poprawę i utrzymanie remisji w ciężkim zaburzeniu depresyjnym.�Dialogi Clin Neurosci.�2008;10(4):377.�[Artykuł bezpłatny PMC][PubMed]
11.�Fekadu A, Wooderson SC, Markopoulou K, Cleare AJ. Metoda Maudsley Staging dla depresji opornej na leczenie: przewidywanie długoterminowego wyniku i utrzymywania się objawów.J Clin Psychiatria.�2009;70(7):952.�[PubMed]
12.�Bennabi D, Aouizerate B, El-Hage W, et al. Czynniki ryzyka oporności na leczenie w depresji jednobiegunowej: przegląd systematyczny.�J Zaburzenia afektu.�2015;171:137.�[PubMed]
13.�Serretti A, Olgiati P, Liebman MN, et al. Kliniczne przewidywanie odpowiedzi przeciwdepresyjnej w zaburzeniach nastroju: liniowe modele wielowymiarowe vs. modele sieci neuronowych.�Psychiatria Res.�2007;152(2):3.[PubMed]
14.�Driessen E, Hollon SD. Terapia poznawczo-behawioralna zaburzeń nastroju: skuteczność, moderatory i mediatory.�Psychiatra Clin North Am.�2010;33(3):537.�[Artykuł bezpłatny PMC][PubMed]
15.�Cleare A, Pariante C, Young A, et al. Członkowie Consensus Meeting opartych na dowodach wytycznych dotyczących leczenia zaburzeń depresyjnych lekami przeciwdepresyjnymi: przegląd wytycznych brytyjskiego stowarzyszenia na rzecz psychofarmakologii z 2008 r.�J Psychopharmacol.�2015;29(5):459.�[PubMed]
16.�Tunnard C, Rane LJ, Wooderson SC, i in. Wpływ niekorzystnych sytuacji w dzieciństwie na samobójstwa i przebieg kliniczny w depresji lekoopornej.�J Zaburzenia afektu.�2014;152:122.�[PubMed]
17.�Nemeroff CB, Heim CM, Thase ME, et al. Różnice w odpowiedziach na psychoterapię a farmakoterapię u pacjentów z przewlekłymi postaciami dużej depresji i dziecięcych urazów.�Proc Natl Acad Sci USA A.�2003;100(24):14293.�[Artykuł bezpłatny PMC][PubMed]
18.�Nierenberg AA. Predyktory odpowiedzi na leki przeciwdepresyjne – ogólne zasady i implikacje kliniczne.�Psychiatra Clin North Am.�2003;26(2):345.�[PubMed]
19.�To ja. Wykorzystanie biomarkerów do przewidywania odpowiedzi na leczenie w przypadku dużego zaburzenia depresyjnego: dowody z wcześniejszych i obecnych badań.�Dialogi Clin Neurosci.�2014;16(4):539.�[Artykuł bezpłatny PMC][PubMed]
20.�Jani BD, McLean G, Nicholl BI, et al. Ocena ryzyka i przewidywanie wyników u pacjentów z objawami depresji: przegląd potencjalnej roli biomarkerów krwi obwodowej.Front Hum Neurosci.�2015;9:18.�[Artykuł bezpłatny PMC][PubMed]
21.�Suravajhala P, Kogelman LJ, Kadarmideen HN. Integracja i analiza danych wieloomowych przy użyciu podejść do genomiki systemów: metody i zastosowania w produkcji zwierzęcej, zdrowiu i dobrostanie.�Genet Sel Evol.�2016;48(1):1.�[Artykuł bezpłatny PMC][PubMed]
22.�Menke A. Ekspresja genów: Biomarker terapii przeciwdepresyjnej?�Int Rev Psychiatria.�2013;25(5):579.�[PubMed]
23.�Peng B, Li H, Peng XX. Metabolomika funkcjonalna: od odkrywania biomarkerów do przeprogramowania metabolomu.�Komórka białkowa.�2015;6(9):628.�[Artykuł bezpłatny PMC][PubMed]
24.�Aagaard K, Petrosino J, Keitel W, et al. Strategia Human Microbiome Project dotycząca kompleksowego pobierania próbek ludzkiego mikrobiomu i dlaczego ma to znaczenie.�FASEB J.�2013;27(3):1012.[Artykuł bezpłatny PMC][PubMed]
25.�Sonner Z, Wilder E, Heikenfeld J, et al. Mikroprzepływy ekrynowego gruczołu potowego, w tym implikacje podziału biomarkerów, transportu i bioczujników.Biomikrofluidyka.�2015;9(3): 031301.[Artykuł bezpłatny PMC][PubMed]
27.�J Brand S, Moller M, H Harvey B. Przegląd biomarkerów w zaburzeniach nastroju i psychotycznych: analiza korelacji klinicznych i przedklinicznych.Curr Neuropharmacol.�2015;13(3):324.[Artykuł bezpłatny PMC][PubMed]
29.�Fu CH, Steiner H, Costafreda SG. Predykcyjne biomarkery neuronalne odpowiedzi klinicznej w depresji: metaanaliza funkcjonalnych i strukturalnych badań neuroobrazowania terapii farmakologicznych i psychologicznych.Neurobiol Dis.�2013;52:75.�[PubMed]
30.�Mamdani F, Berlim M, Beaulieu M, Labbe A, Merette C, Turecki G. Biomarkery ekspresji genów odpowiedzi na leczenie citalopramem w ciężkim zaburzeniu depresyjnym.Transl Psychiatria.�2011;1(6): e13.[Artykuł bezpłatny PMC][PubMed]
31.�Smith RS. Teoria makrofagów depresji.�Hipotezy Med.�1991;35(4):298.�[PubMed]
32.�Irwin MR, Miller AH. Zaburzenia depresyjne i odporność: 20 lat postępu i odkrycia.�Odporność na zachowanie mózgu.�2007;21(4):374.�[PubMed]
33.�Maes M, Leonard B, Myint A, Kubera M, Verkerk R. Nowa hipoteza depresji „5-HT”: komórkowa aktywacja immunologiczna indukuje 2,3-dioksygenazę indoloaminy, co prowadzi do obniżenia poziomu tryptofanu w osoczu i zwiększonej syntezy szkodliwe katabolity tryptofanu (TRYCAT), z których oba przyczyniają się do wystąpienia depresji.Prog Neuropsychopharmacol Biol Psychiatry.�2011;35(3):702.[PubMed]
34.�Miller AH, Maletic V, Raison CL. Zapalenie i jego niezadowolenie: Rola cytokin w patofizjologii dużej depresji.�Biol Psychiatria.�2009;65(9):732.�[Artykuł bezpłatny PMC][PubMed]
35.�Miller AH, Raison CL. Rola stanu zapalnego w depresji: od konieczności ewolucyjnej do nowoczesnego celu leczenia.�Nat Rev Immun2016;16(1):22.�[Artykuł bezpłatny PMC][PubMed]
36.�Raison CL, Capuron L, Miller AH. Cytokiny śpiewają blues: stan zapalny i patogenezę depresji.�Trendy Odporność.�2006;27(1):24.�[Artykuł bezpłatny PMC][PubMed]
37.�Raison CL, Felger JC, Miller AH. Odporność na stany zapalne i leczenie w poważnej depresji: idealna burza.�Psychiatr Times.�2013;30(9)
38.�Dowlati Y, Herrmann N, Swardfager W, et al. Metaanaliza cytokin w ciężkiej depresji.�Biol Psychiatria.�2010;67(5):446.�[PubMed]
39.�Eyre HA, Air T, Pradhan A, et al. Metaanaliza chemokin w ciężkiej depresji.�Prog Neuropsychopharmacol Biol Psychiatry.�2016;68:1.�[Artykuł bezpłatny PMC][PubMed]
40.�Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivim�ki M. Zbiorcza metaanaliza interleukin 6 i 1?, czynnik martwicy nowotworu? i białka C-reaktywnego u pacjentów z ciężkimi zaburzeniami depresyjnymi.�Odporność na zachowanie mózgu.�2015;49:206.�[Artykuł bezpłatny PMC][PubMed]
41.�Howren MB, Lamkin DM, Suls J. Powiązania depresji z białkiem C-reaktywnym, IL-1 i IL-6: metaanaliza.Psychosom Med.�2009;71(2):171.�[PubMed]
42.�Liu Y, Ho RC-M, Mak A. Interleukina (IL)-6, czynnik martwicy nowotworu alfa (TNF-a) i rozpuszczalne receptory interleukiny-2 (sIL-2R) są podwyższone u pacjentów z dużym zaburzeniem depresyjnym: meta- analiza i metaregresja.�J Zaburzenia afektu.�2012;139(3):230.�[PubMed]
43.�Strawbridge R, Arnone D, Danese A, Papadopoulos A, Herane Vives A, Cleare AJ. Zapalenie i odpowiedź kliniczna na leczenie w depresji: metaanaliza.�Eur Neuropsychofarmakol.�2015;25(10):1532.�[PubMed]
44.�Farooq RK, Asghar K, Kanwal S, Zulqernain A. Rola cytokin zapalnych w depresji: Skoncentruj się na interleukinie-1? (Recenzja)�Przedstawiciel Biomedu�2017;6(1):15.�[Artykuł bezpłatny PMC][PubMed]
45.�Cattaneo A, Ferrari C, Uher R, et al. Pomiary bezwzględne czynnika hamującego migrację makrofagów i interleukiny-1-? Poziomy mRNA dokładnie przewidują odpowiedź na leczenie u pacjentów z depresją.�Int J Neuropsychopharmacol.�2016;19(10):pyw045.�[Artykuł bezpłatny PMC][PubMed]
46.�Baune B, Smith E, Reppermund S, et al. Biomarkery stanu zapalnego przewidują objawy depresyjne, ale nie lękowe podczas starzenia: prospektywne badanie pamięci Sydney i starzenia się.Psychoneuroendokrynol.�2012;37(9):1521.�[PubMed]
47.�Fornaro M, Rocchi G, Escelsior A, Contini P, Martino M. Różne trendy w cytokinach u pacjentów z depresją otrzymujących duloksetynę mogą wskazywać na różne podłoże biologiczne.J Zaburzenia afektu.�2013;145(3):300.�[PubMed]
48.�Hernandez ME, Mendieta D, Martinez-Fong D, et al. Różnice w poziomach krążących cytokin podczas 52-tygodniowego cyklu leczenia SSRI w przypadku dużego zaburzenia depresyjnego.�Eur Neuropsychofarmakol.�2008;18(12):917.�[PubMed]
49.�Hannestad J, DellaGioia N, Bloch M. Wpływ leczenia lekami przeciwdepresyjnymi na poziom cytokin zapalnych w surowicy: metaanaliza.Neuropsychofarmakologia.�2011;36(12):2452.[Artykuł bezpłatny PMC][PubMed]
50.�Hiles SA, Attia J, Baker AL. Zmiany w interleukinie-6, białku C-reaktywnym i interleukinie-10 u osób z depresją po leczeniu przeciwdepresyjnym: metaanaliza.Brain Behav Immun; Prezentacja na: 17. dorocznym spotkaniu Towarzystwa Badawczego PsychoNeuroImmunologii PsychoNeuroImmunology: Crossing Disciplines to Combat Disease; 2012. s. S44.
51.�Harley J, Luty S, Carter J, Mulder R, Joyce P. Podwyższone białko C-reaktywne w depresji: predyktor dobrego długoterminowego wyniku z lekami przeciwdepresyjnymi i złego wyniku z psychoterapią.J Psychopharmacol.�2010;24(4):625.�[PubMed]
52.�Uher R, Tansey KE, Dew T, et al. Biomarker stanu zapalnego jako różnicujący predyktor wyniku leczenia depresji escitalopramem i nortryptyliną.Jestem J Psychiatrią.�2014;171(2):1278.[PubMed]
53.�Chang HH, Lee IH, Gean PW i in. Odpowiedź na leczenie i upośledzenie funkcji poznawczych w ciężkiej depresji: związek z białkiem C-reaktywnym.�Odporność na zachowanie mózgu.�2012;26(1):90.�[PubMed]
54.�Raison CL, Rutherford RE, Woolwine BJ, et al. Randomizowane kontrolowane badanie antagonisty czynnika martwicy nowotworu, infliksymabu, w depresji opornej na leczenie: rola podstawowych biomarkerów stanu zapalnego.JAMA Psychiatria.�2013;70(1):31.�[Artykuł bezpłatny PMC][PubMed]
55.�Krishnadas R, Cavanagh J. Depresja: choroba zapalna?�J Neurol Neurosurg Psychiatria.�2012;83(5):495.�[PubMed]
56.�Raison CL, Miller AH. Czy depresja jest chorobą zapalną?�Curr Psychiatry Rep.�2011;13(6):467.�[Artykuł bezpłatny PMC][PubMed]
57.�Simon N, McNamara K, Chow C, et al. Szczegółowe badanie nieprawidłowości cytokin w przebiegu ciężkiej depresji.�Eur Neuropsychofarmakol.�2008;18(3):230.�[Artykuł bezpłatny PMC][PubMed]
58.�Dahl J, Ormstad H, Aass HC i in. Poziomy różnych cytokin w osoczu wzrastają podczas trwającej depresji i zmniejszają się do normalnego poziomu po wyzdrowieniu.�Psychoneuroendokrynol.�2014;45:77.�[PubMed]
59.�Stelzhammer V, Haenisch F, Chan MK i in. Zmiany proteomiczne w surowicy pacjentów z ciężką depresją nie leczonych wcześniej lekami przeciwdepresyjnymi.�Int J Neuropsychopharmacol.�2014;17(10):1599.�[PubMed]
60.�Liu Y, HO RCM, Mak A. Rola interleukiny (IL)-17 w lęku i depresji pacjentów z reumatoidalnym zapaleniem stawów.Int J Rheum Dis.�2012;15(2):183.�[PubMed]
61.�Diniz BS, Sibille E, Ding Y, et al. Biosygnatura osocza i patologia mózgu związana z uporczywym upośledzeniem funkcji poznawczych w depresji późnego życia.�Mol Psychiatria.�2015;20(5):594.�[Artykuł bezpłatny PMC][PubMed]
62.�Janelidze S, Ventorp F, Erhardt S, et al. Zmienione poziomy chemokin w płynie mózgowo-rdzeniowym i osoczu osób próbujących samobójstwoPsychoneuroendokrynol.�2013;38(6):853.�[PubMed]
63.�Powell TR, Schalkwyk LC, Heffernan AL i in. Czynnik martwicy nowotworu i jego cele w szlaku cytokin zapalnych są identyfikowane jako przypuszczalne biomarkery transkryptomiczne odpowiedzi na escitalopram.Eur Neuropsychofarmakol.�2013;23(9):1105.�[PubMed]
64.�Wong M, Dong C, Maestre-Mesa J, Licinio J. Polimorfizmy w genach związanych ze stanem zapalnym są związane z podatnością na poważną depresję i reakcją przeciwdepresyjną.Mol Psychiatria.�2008;13(8):800.�[Artykuł bezpłatny PMC][PubMed]
65.�Kling MA, Alesci S, Csako G, et al. Utrzymujący się stan prozapalny niskiego stopnia u nieleczonych, zremisowanych kobiet z poważnymi zaburzeniami depresyjnymi, o czym świadczy podwyższone stężenie białek ostrej fazy białka C-reaktywnego i amyloidu A w surowicy.Biol Psychiatria.�2007;62(4):309.�[Artykuł bezpłatny PMC][PubMed]
66.�Schaefer M, Sarkar S, Schwarz M, Friebe A. Rozpuszczalna wewnątrzkomórkowa cząsteczka adhezyjna-1 u pacjentów z jednobiegunowymi lub dwubiegunowymi zaburzeniami afektywnymi: wyniki z badania pilotażowego.Neuropsychobiol.�2016;74(1):8.[PubMed]
67.�Dimopoulos N, Piperi C, Salonicioti A, et al. Podwyższenie stężenia cząsteczek adhezyjnych w osoczu w depresji w późnym okresie życia.�Int J Geriatr Psychiatria.�2006;21(10):965.�[PubMed]
68.�Bocchio-Chiaveto L, Bagnardi V, Zanardini R, et al. Poziomy BDNF w surowicy i osoczu w ciężkiej depresji: badanie replikacji i metaanalizy.�Świat J Biol Psychiatry.�2010;11(6):763.�[PubMed]
69.�Brunoni AR, Lopes M, Fregni F. Przegląd systematyczny i metaanaliza badań klinicznych dotyczących dużej depresji i poziomów BDNF: implikacje dla roli neuroplastyczności w depresji.Int J Neuropsychopharmacol.�2008;11(8):1169.�[PubMed]
70.�Molendijk M, Spinhoven P, Polak M, Bus B, Penninx B, Elzinga B. Stężenia BDNF w surowicy jako obwodowe objawy depresji: dowody z przeglądu systematycznego i metaanaliz 179 skojarzeń.Mol Psychiatria.�2014;19(7):791.�[PubMed]
71.�Sen S, Duman R, Sanacora G. Surowiczy czynnik neurotroficzny pochodzenia mózgowego, depresja i leki przeciwdepresyjne: metaanalizy i implikacje.Biol Psychiatria.�2008;64(6):527.�[Artykuł bezpłatny PMC][PubMed]
72.�Zhou L, Xiong J, Lim Y, et al. Regulacja w górę proBDNF we krwi i jego receptorów w ciężkiej depresji.�J Zaburzenia afektu.�2013;150(3):776.�[PubMed]
73.�Chen YW, Lin PY, Tu KY, Cheng YS, Wu CK, Tseng PT. Znacząco niższy poziom czynnika wzrostu nerwów u pacjentów z poważnymi zaburzeniami depresyjnymi niż u zdrowych osób: metaanaliza i przegląd systematyczny.Leczenie neuropsychiatryczne.�2014;11:925.�[Artykuł bezpłatny PMC][PubMed]
74.�Lin PY, Tseng PT. Zmniejszone poziomy czynnika neurotroficznego pochodzącego z linii komórek glejowych u pacjentów z depresją: badanie metaanalityczne.J Psychiatr Res.�2015;63:20.�[PubMed]
75.�Warner-Schmidt JL, Duman RS. VEGF jako potencjalny cel interwencji terapeutycznej w depresji.�Curr Op Pharmacol.�2008;8(1):14.�[Artykuł bezpłatny PMC][PubMed]
76.�Carvalho AF, K.hler CA, McIntyre RS, et al. Czynnik wzrostu śródbłonka naczyń obwodowych jako nowy biomarker depresji: metaanaliza.�Psychoneuroendokrynol.�2015;62:18.�[PubMed]
77.�Tseng PT, Cheng YS, Chen YW, Wu CK, Lin PY. Podwyższony poziom czynnika wzrostu śródbłonka naczyniowego u pacjentów z poważnymi zaburzeniami depresyjnymi: metaanaliza.Eur Neuropsychofarmakol.�2015;25(10):1622.�[PubMed]
78.�Carvalho L, Torre J, Papadopoulos A, et al. Brak klinicznych korzyści terapeutycznych ze stosowania leków przeciwdepresyjnych wiąże się z ogólną aktywacją układu zapalnego.�J Zaburzenia afektu.�2013;148(1):136.�[PubMed]
79.�Clark-Raymond A, Meresh E, Hoppensteadt D, et al. Czynnik wzrostu śródbłonka naczyniowego: potencjalny predyktor odpowiedzi na leczenie w przypadku dużej depresji.�Świat J Biol Psychiatry.�2015: 1.�[PubMed]
80.�Isung J, Mobarrez F, Nordstr.m P, sberg M, Jokinen J. Niski czynnik wzrostu śródbłonka naczyniowego w osoczu (VEGF) związany z dokonanym samobójstwem.Świat J Biol Psychiatry.�2012;13(6):468.�[PubMed]
81.�Buttensch�n HN, Foldager L, Elfving B, Poulsen PH, Uher R, Mors O. Czynniki neurotroficzne w depresji w odpowiedzi na leczenie.J Zaburzenia afektu.�2015;183:287.�[PubMed]
82.�Szczęsny E, Ślusarczyk J, Głombik K, et al. Możliwy udział IGF-1 w zaburzeniach depresyjnych.�Przedstawiciel Pharmacol�2013;65(6):1622.�[PubMed]
83.�Tu KY, Wu MK, Chen YW i in. Istotnie wyższe poziomy obwodowego insulinopodobnego czynnika wzrostu-1 u pacjentów z dużym zaburzeniem depresyjnym lub zaburzeniem afektywnym dwubiegunowym niż u zdrowych osób z grupy kontrolnej: metaanaliza i przegląd w wytycznych PRISMA.Med.�2016;95(4):e2411.�[Artykuł bezpłatny PMC][PubMed]
84.�Wu CK, Tseng PT, Chen YW, Tu KY, Lin PY. Znacząco wyższy poziom obwodowego czynnika wzrostu fibroblastów-2 u pacjentów z poważnymi zaburzeniami depresyjnymi: wstępna metaanaliza zgodnie z wytycznymi MOOSE.Med.�2016;95(33):e4563.�[Artykuł bezpłatny PMC][PubMed]
85.�He S, Zhang T, Hong B, et al. Zmniejszone poziomy czynnika wzrostu fibroblastów w surowicy-2 u pacjentów z poważnymi zaburzeniami depresyjnymi przed i po leczeniu.Neurosci Lett.�2014;579:168.�[PubMed]
86.�Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN. Zmieniona ekspresja genów czynnika neurotroficznego pochodzenia mózgowego i receptorowej kinazy tyrozynowej B w mózgu pośmiertnych osobników samobójczych.Psychiatria Arch Gen.�2003;60(8):804.�[PubMed]
87.�Srikanthan K, Feyh A, Visweshwar H, Shapiro JI, Sodhi K. Systematyczny przegląd biomarkerów zespołu metabolicznego: panel do wczesnego wykrywania, zarządzania i stratyfikacji ryzyka w populacji Zachodniej Wirginii.Int J Med Sci.�2016;13(1):25.�[Artykuł bezpłatny PMC][PubMed]
88.�Lu XY. Hipoteza leptynowa depresji: potencjalny związek między zaburzeniami nastroju a otyłością?�Curr Op Pharmacol.�2007;7(6):648.�[Artykuł bezpłatny PMC][PubMed]
89.�Wittekind DA, Kluge M. Ghrelin w zaburzeniach psychiatrycznych� Przegląd.�Psychoneuroendokrynol.�2015;52:176.�[PubMed]
90.�Kan C, Silva N, Golden SH, et al. Przegląd systematyczny i metaanaliza związku między depresją a insulinoopornością.�Opieka cukrzycowa.�2013;36(2):480.�[Artykuł bezpłatny PMC][PubMed]
91.�Liu X, Li J, Zheng P, et al. Lipidomika osocza ujawnia potencjalne markery lipidowe poważnego zaburzenia depresyjnego.�Anal Bioanal Chem.�2016;408(23):6497.�[PubMed]
92.�Lustman PJ, Anderson RJ, Freedland KE, De Groot M, Carney RM, Clouse RE. Depresja i słaba kontrola glikemii: metaanalityczny przegląd literatury.�Opieka cukrzycowa.�2000;23(7):934.�[PubMed]
93.�Maes M. Dowody na odpowiedź immunologiczną w ciężkiej depresji: przegląd i hipoteza.�Prog NeuroPsychopharmacol Biol Psychiatry.�1995;19(1):11.�[PubMed]
94.�Zheng H, Zheng P, Zhao L i in. Diagnostyka predykcyjna ciężkiej depresji przy użyciu metabolomiki opartej na NMR i maszyny wektorów wspierających metodą najmniejszych kwadratów.�Clinica Chimica Acta.�2017;464: 223 227.[PubMed]
95.�Xia Q, Wang G, Wang H, Xie Z, Fang Y, Li Y. Badanie metabolizmu glukozy i lipidów u pacjentów z depresją pierwszego epizodu.J Clin Psychiatria.�2009;19: 241 243.
97.�Jacobsen JP, Krystal AD, Krishnan KRR, Caron MG. Wspomaganie o powolnym uwalnianiu 5-hydroksytryptofanu w leczeniu depresji opornej na leczenie: uzasadnienie kliniczne i przedkliniczne.Trendy Pharmacol Sci.�2016;37(11):933.�[Artykuł bezpłatny PMC][PubMed]
98.�Salamone JD, Correa M, Yohn S, Cruz LL, San Miguel N, Alatorre L. Farmakologia zachowań związanych z wyborem związanym z wysiłkiem: dopamina, depresja i różnice indywidualne.Zachowanie procesów.�2016;127:3.�[PubMed]
99.�Coplan JD, Gopinath S, Abdallah CG, Berry BR. Neurobiologiczna hipoteza o opornej na leczenie depresji.Mechanizmy nieskuteczności selektywnego inhibitora wychwytu zwrotnego serotoniny.Przód Zachowanie Neurosci.�2014;8:189.�[Artykuł bezpłatny PMC][PubMed]
100.�Popa D, Cerdan J, Rep�rant C, et al. Podłużne badanie odpływu 5-HT podczas przewlekłego leczenia fluoksetyną przy użyciu nowej techniki przewlekłej mikrodializy u wysoce emocjonalnego szczepu myszy.Eur J Pharmacol.�2010;628(1):83.�[PubMed]
101.�Atake K, Yoshimura R, Hori H, et al. Duloksetyna, selektywny inhibitor wychwytu zwrotnego noradrenaliny, zwiększała stężenie 3-metoksy-4-hydroksyfenyloglikolu w osoczu, ale nie kwasu homowanilowego u pacjentów z ciężkimi zaburzeniami depresyjnymi.Clin Psychopharmacol Neurosci.�2014;12(1):37.�[Artykuł bezpłatny PMC][PubMed]
102.�Ueda N, Yoshimura R, Shinkai K, Nakamura J. Poziomy metabolitów katecholamin w osoczu przewidują odpowiedź na sulpiryd lub fluwoksaminę w ciężkiej depresji.Farmakopsychiatria.�2002;35(05):175.[PubMed]
103.�Yamana M, Atake K, Katsuki A, Hori H, Yoshimura R. Biologiczne markery krwi do przewidywania odpowiedzi na escitalopram u pacjentów z poważnymi zaburzeniami depresyjnymi: badanie wstępne.J Depresja niepokoju.�2016;5: 222.
104.�Parker KJ, Schatzberg AF, Lyons DM. Neuroendokrynne aspekty hiperkortyzolizmu w ciężkiej depresji.�Zachowanie Horm.�2003;43(1):60.�[PubMed]
105.�Stetlera C, Millera GE. Depresja i aktywacja podwzgórze-przysadka-nadnercza: ilościowe podsumowanie czterech dekad badań.Psychosom Med.�2011;73(2):114.�[PubMed]
106.�Herane Vives A, De Angel V, Papadopoulos A, et al. Związek między kortyzolem, stresem i chorobą psychiczną: nowe spostrzeżenia dzięki analizie włosów.�J Psychiatr Res.�2015;70:38.�[PubMed]
107.�Fischer S, Strawbridge R, Vives AH, Cleare AJ. Kortyzol jako predyktor odpowiedzi na terapię psychologiczną w zaburzeniach depresyjnych: przegląd systematyczny i metaanaliza.�Br J Psychiatria.�2017;210(2):105.�[PubMed]
108.�Anacker C, Zunszain PA, Carvalho LA, Pariante CM. Receptor glukokortykoidowy: oś depresji i leczenia przeciwdepresyjnego?�Psychoneuroendokrynologia.�2011;36(3):415.�[Artykuł bezpłatny PMC][PubMed]
109.�Markopoulou K, Papadopoulos A, Juruena MF, Poon L, Pariante CM, Cleare AJ. Stosunek kortyzolu do DHEA w depresji opornej na leczenie.�Psychoneuroendokrynol.�2009;34(1):19.�[PubMed]
110.�Joffe RT, Pearce EN, Hennessey JV, Ryan JJ, Stern RA. Subkliniczna niedoczynność tarczycy, nastrój i funkcje poznawcze u osób starszych: przegląd.�Int J Geriatr Psychiatria.�2013;28(2):111.�[Artykuł bezpłatny PMC][PubMed]
111.�Duval F, Mokrani MC, Erb A, et al. Chronobiologiczny stan osi podwzgórzowo-przysadkowo-tarczycowej i działanie przeciwdepresyjne w ciężkiej depresji.�Psychoneuroendokrynol.�2015;59:71.�[PubMed]
112.�Marsden W. Plastyczność synaptyczna w depresji: korelaty molekularne, komórkowe i funkcjonalne.�Prog Neuropsychopharmacol Biol Psychiatry.�2013;43:168.�[PubMed]
113.�Duman RS, Voleti B. Ścieżki sygnałowe leżące u podstaw patofizjologii i leczenia depresji: nowe mechanizmy dla szybko działających środków.Trendy Neurosci.�2012;35(1):47.[Artykuł bezpłatny PMC][PubMed]
114.�Ripke S, Wray NR, Lewis CM i in. Mega-analiza badań asocjacyjnych całego genomu dla poważnych zaburzeń depresyjnych.�Mol Psychiatria.�2013;18(4):497.�[Artykuł bezpłatny PMC][PubMed]
115.�Mullins N, Power R, Fisher H, et al. Interakcje wielogenowe z niekorzystnymi warunkami środowiskowymi w etiologii dużej depresji.�Psychol Med.�2016;46(04):759.�[Artykuł bezpłatny PMC][PubMed]
116.�Lewis S. Zaburzenia neurologiczne: telomery i depresja.�Nat Rev Neurosci.�2014;15(10): 632.[PubMed]
117.�Lindqvist D, Epel ES, Mellon SH i in. Zaburzenia psychiczne i długość telomerów leukocytów: podstawowe mechanizmy łączące choroby psychiczne ze starzeniem się komórek.Neurosci Biobehav Rev.�2015;55:333.�[Artykuł bezpłatny PMC][PubMed]
118.�McCall WV. Biomarker aktywności spoczynkowej do przewidywania odpowiedzi na SSRI w ciężkich zaburzeniach depresyjnych.�J Psychiatr Res.�2015;64:19.�[Artykuł bezpłatny PMC][PubMed]
119.�Schuch FB, Deslandes AC, Stubbs B, Gosmann NP, da Silva CTB, de Almeida Fleck MP. Neurobiologiczny wpływ ćwiczeń na ciężkie zaburzenie depresyjne: przegląd systematyczny.�Neurosci Biobehav Rev.�2016;61:1.�[PubMed]
120.�Foster JA, Neufeld K-AM. Oś jelitowo-mózgowa: jak mikrobiom wpływa na lęk i depresjęTrendy Neurosci.�2013;36(5):305.�[PubMed]
121.�Quattrocki E, Baird A, Yurgelun-Todd D. Biologiczne aspekty związku między paleniem a depresją.Harv Rev Psychiatria.�2000;8(3):99.�[PubMed]
122.�Maes M, Kubera M, Obuchowiczwa E, Goehler L, Brzeszcz J. Wiele chorób współistniejących depresji wyjaśnione przez szlaki stresu (neuro)zapalnego oraz oksydacyjnego i nitrosatywnego.Neuro Endocrinol Lett.�2011;32(1):7.�[PubMed]
123.�Miller G, Rohleder N, Cole SW. Przewlekły stres interpersonalny przewiduje aktywację pro- i przeciwzapalnych szlaków sygnalizacyjnych sześć miesięcy później.�Psychosom Med.�2009;71(1):57.�[Artykuł bezpłatny PMC][PubMed]
124.�Steptoe A, Hamer M, Chida Y. Wpływ ostrego stresu psychologicznego na krążące czynniki zapalne u ludzi: przegląd i metaanaliza.Odporność na zachowanie mózgu.�2007;21(7):901.�[PubMed]
125.�Danese A, Moffitt TE, Harrington H i in. Niekorzystne doświadczenia z dzieciństwa i czynniki ryzyka dla dorosłych dla chorób związanych z wiekiem: depresja, stany zapalne i grupowanie markerów ryzyka metabolicznego.Arch Pediatr Adolesc Med.�2009;163(12):1135.�[Artykuł bezpłatny PMC][PubMed]
126.�Danese A, Pariante CM, Caspi A, Taylor A, Poulton R. Maltretowanie w dzieciństwie przewiduje stan zapalny u dorosłych w badaniu przebiegu życia.Proc Natl Acad Sci USA A.�2007;104(4):1319.�[Artykuł bezpłatny PMC][PubMed]
127.�Danese A, Caspi A, Williams B, et al. Biologiczne osadzenie stresu poprzez procesy zapalne w dzieciństwie.�Mol Psychiatria.�2011;16(3):244.�[Artykuł bezpłatny PMC][PubMed]
128.�Suzuki A, Poon L, Kumari V, Cleare AJ. Błędy lękowe w emocjonalnym przetwarzaniu twarzy po traumie z dzieciństwa jako wskaźnik odporności i podatności na depresję.�Maltretowanie dziecka.�2015;20(4):240.�[PubMed]
129.�Strawbridge R, młody AH. Oś HPA i dysregulacja poznawcza w zaburzeniach nastroju. W: McIntyre RS, Cha DS, redaktorzy.�Zaburzenia funkcji poznawczych w dużych zaburzeniach depresyjnych: znaczenie kliniczne, podłoża biologiczne i możliwości leczenia.�Cambridge: Cambridge University Press; 2016. s. 179.
130.�Keller J, Gomez R, Williams G, et al. Oś HPA w ciężkiej depresji: kortyzol, symptomatologia kliniczna i zmienność genetyczna przewidują funkcje poznawcze.�Mol Psychiatria.�2016 sierpnia 16;�Epub.�[Artykuł bezpłatny PMC][PubMed]
132.�Chen Y, Baram TZ. W kierunku zrozumienia, w jaki sposób stres we wczesnym okresie życia przeprogramowuje kognitywne i emocjonalne sieci mózgowe.�Neuropsychofarmakol.�2015;41(1):197.�[Artykuł bezpłatny PMC][PubMed]
133.�Porter RJ, Gallagher P, Thompson JM, Young AH. Zaburzenia neurokognitywne u pacjentów nieprzyjmujących leków z ciężkimi zaburzeniami depresyjnymi.�Br J Psychiatria.�2003;182:214.�[PubMed]
134.�Gallagher P, Robinson L, Gray J, Young A, Porter R. Funkcja neurokognitywna po remisji w ciężkim zaburzeniu depresyjnym: potencjalny obiektywny marker odpowiedzi?Aust NZJ Psychiatria.�2007;41(1):54.�[PubMed]
135.�Pittenger C, Duman RS. Stres, depresja i neuroplastyczność: zbieżność mechanizmów.�Neuropsychofarmakol.�2008;33(1):88.�[PubMed]
136.�B�ckman L, Nyberg L, Lindenberger U, Li SC, Farde L. Korelacyjna triada między starzeniem się, dopaminą i poznaniem: obecny stan i perspektywy na przyszłość.Neurosci Biobehav Rev.�2006;30(6):791.�[PubMed]
137.�Allison DJ, Ditor DS. Powszechna etiologia zapalna depresji i zaburzeń poznawczych: cel terapeutyczny.�J Neurozapalenie.�2014;11:151.�[Artykuł bezpłatny PMC][PubMed]
138.�Rosenblat JD, Brietzke E, Mansur RB, Maruschak NA, Lee Y, McIntyre RS. Zapalenie jako neurobiologiczne podłoże upośledzenia funkcji poznawczych w chorobie afektywnej dwubiegunowej: dowody, patofizjologia i implikacje leczenia.J Zaburzenia afektu.�2015;188:149.�[PubMed]
139.�Krogh J, Benros ME, Jrgensen MB, Vesterager L, Elfving B, Nordentoft M. Związek między objawami depresyjnymi, funkcjami poznawczymi i stanem zapalnym w ciężkiej depresji.Odporność na zachowanie mózgu.�2014;35:70.�[PubMed]
140.�Soares CN, Zitek B. Wrażliwość na hormony reprodukcyjne i ryzyko depresji w cyklu życia kobiety: kontinuum wrażliwości?J Psychiatria Neurosci.�2008;33(4):331.�[Artykuł bezpłatny PMC][PubMed]
141.�Hiles SA, Baker AL, de Malmanche T, Attia J. Metaanaliza różnic w IL-6 i IL-10 między osobami z depresją i bez: badanie przyczyn niejednorodności.Odporność na zachowanie mózgu.�2012;26(7):1180.�[PubMed]
142.�Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S. Wydzielanie adipokiny trzewnej tłuszczu jest związane z ogólnoustrojowym zapaleniem u otyłych ludzi.Cukrzyca.�2007;56(4):1010.�[PubMed]
143.�Divani AA, Luo X, Datta YH, Flaherty JD, Panoskaltsis-Mortari A. Wpływ doustnych i dopochwowych hormonalnych środków antykoncepcyjnych na zapalne biomarkery krwi.Mediatorzy Zapalenie.�2015;2015: 379501.[Artykuł bezpłatny PMC][PubMed]
144.�Ramsey JM, Cooper JD, Penninx BW, Bahn S. Zmienność biomarkerów w surowicy z płcią i statusem hormonalnym kobiet: implikacje dla testów klinicznych.Przedstawiciel naukowy�2016;6:26947.�[Artykuł bezpłatny PMC][PubMed]
145.�Eyre H, Lavretsky H, Kartika J, Qassim A, Baune B. Modulacyjne działanie klas leków przeciwdepresyjnych na wrodzony i adaptacyjny układ odpornościowy w depresji.Farmakopsychiatria.�2016;49(3):85.[Artykuł bezpłatny PMC][PubMed]
146.�Hiles SA, Baker AL, de Malmanche T, Attia J. Interleukina-6, białko C-reaktywne i interleukina-10 po leczeniu przeciwdepresyjnym u osób z depresją: metaanaliza.Psychol Med.�2012;42(10):2015.�[PubMed]
147.�Janssen DG, Caniato RN, Verster JC, Baune BT. Przegląd psychoneuroimmunologiczny dotyczący cytokin zaangażowanych w odpowiedź na leczenie przeciwdepresyjne.�Hum Psychopharmacol.�2010;25(3):201.�[PubMed]
148.�Artigas F. Receptory serotoninowe zaangażowane w działanie przeciwdepresyjne.�Pharmacol Ther.�2013;137(1):119.�[PubMed]
149.�Lee BH, Kim YK. Rola BDNF w patofizjologii dużej depresji oraz w leczeniu przeciwdepresyjnym.�Badanie psychiatryczne.�2010;7(4):231.�[Artykuł bezpłatny PMC][PubMed]
150.�Hashimoto K. Zapalne biomarkery jako zróżnicowane predyktory odpowiedzi przeciwdepresyjnej.�Int J Mol Sci.�2015;16(4):7796.�[Artykuł bezpłatny PMC][PubMed]
151.�Goldberg D. Heterogeniczność „dużej depresji”�Psychiatria Świata.�2011;10(3):226.[Artykuł bezpłatny PMC][PubMed]
152.�Arnow BA, Blasey C, Williams LM i in. Podtypy depresji w przewidywaniu odpowiedzi na leki przeciwdepresyjne: raport z badania iSPOT-D.�Jestem J Psychiatrią.�2015;172(8):743.�[PubMed]
153.�Kunugi H, Hori H, Ogawa S. Markery biochemiczne podtypujące poważne zaburzenie depresyjne.Psychiatria Clin Neurosci.�2015;69(10):597.�[PubMed]
154.�Baune B, Stuart M, Gilmour A, et al. Związek między podtypami depresji a chorobami układu krążenia: systematyczny przegląd modeli biologicznych.�Transl Psychiatria.�2012;2(3): e92.[Artykuł bezpłatny PMC][PubMed]
155.�Vogelzangs N, Duivis HE, Beekman AT, et al. Związek zaburzeń depresyjnych, cech depresji i leków przeciwdepresyjnych ze stanem zapalnym.�Transl Psychiatria.�2012;2: e79.[Artykuł bezpłatny PMC][PubMed]
156.�Lamers F, Vogelzangs N, Merikangas K, De Jonge P, Beekman A, Penninx B. Dowody na różną rolę funkcji osi HPA, zapalenia i zespołu metabolicznego w depresji melancholijnej i atypowej.Mol Psychiatria.�2013;18(6):692.�[PubMed]
157.�Penninx BW, Milaneschi Y, Lamers F, Vogelzangs N. Zrozumienie somatycznych konsekwencji depresji: mechanizmy biologiczne i rola profilu objawów depresji.BMC Med.�2013;11(1): 1.[Artykuł bezpłatny PMC][PubMed]
158.�Capuron L, Su S, Miller AH, et al. Objawy depresji i zespół metaboliczny: czy stan zapalny jest podstawowym ogniwem?�Biol Psychiatria.�2008;64(10):896.�[Artykuł bezpłatny PMC][PubMed]
159.�Dantzer R, O�Connor JC, Freund GG, Johnson RW, Kelley KW. Od stanu zapalnego po choroby i depresję: kiedy układ odpornościowy ujarzmia mózg.�Nat Rev Neurosci.�2008;9(1):46.[Artykuł bezpłatny PMC][PubMed]
160.�Maes M, Berk M, Goehler L, et al. Zachowania depresyjne i chorobowe to reakcje typu Janus na wspólne szlaki zapalneBMC Med.�2012;10:66.�[Artykuł bezpłatny PMC][PubMed]
161.�Merikangas KR, Jin R, He JP, et al. Rozpowszechnienie i korelaty zaburzeń afektywnych dwubiegunowych w światowej inicjatywie badania zdrowia psychicznego.�Psychiatria Arch Gen.�2011;68(3):241.�[Artykuł bezpłatny PMC][PubMed]
162.�Hirschfeld RM, Lewis L, Vornik LA. Postrzeganie i wpływ choroby afektywnej dwubiegunowej: jak daleko naprawdę zaszliśmy? Wyniki ankiety przeprowadzonej wśród osób z chorobą afektywną dwubiegunową z 2000 r. przeprowadzonej przez ogólnokrajowe stowarzyszenie depresyjne i maniakalno-depresyjne.�J Clin Psychiatria.�2003;64(2):161.�[PubMed]
163.�Young AH, MacPherson H. Wykrywanie choroby afektywnej dwubiegunowej.�Br J Psychiatria.�2011;199(1):3.[PubMed]
164.�V.hringer PA, Perlis RH. Rozróżnianie między chorobą afektywną dwubiegunową a dużą depresją.�Psychiatra Clin North Am.�2016;39(1):1.�[PubMed]
165.�Becking K, Spijker AT, Hoencamp E, Penninx BW, Schoevers RA, Boschloo L. Zaburzenia w osi podwzgórze-przysadka-nadnercza i aktywność immunologiczna różnicująca epizody depresyjne jednobiegunowe i dwubiegunowe.PLoS Jeden.�2015;10(7):e0133898.�[Artykuł bezpłatny PMC][PubMed]
166.�Huang TL, Lin FC. Wysokowrażliwe poziomy białka C-reaktywnego u pacjentów z ciężkimi zaburzeniami depresyjnymi i manią dwubiegunową.�Prog NeuroPsychopharmacol Biol Psychiatry.�2007;31(2):370.�[PubMed]
167.�Angst J, Gamma A, Endrass J. Czynniki ryzyka dla widm dwubiegunowych i depresyjnych.�Acta Psychiatr Scand.�2003;418:15.�[PubMed]
168.�Fekadu A, Wooderson S, Donaldson C, et al. Wielowymiarowe narzędzie do ilościowej oceny oporności na leczenie w depresji: metoda inscenizacji Maudsleya.�J Clin Psychiatria.�2009;70(2):177.�[PubMed]
169.�Papakostas G, Shelton R, Kinrys G, et al. Ocena wielotestowego biologicznego testu diagnostycznego opartego na surowicy dla poważnych zaburzeń depresyjnych: badanie pilotażowe i replikacyjne.Mol Psychiatria.�2013;18(3):332.�[PubMed]
170.�Fan J, Han F, Liu H. Wyzwania analizy dużych zbiorów danych.�Natl Sci Rev.�2014;1(2):293.[Artykuł bezpłatny PMC][PubMed]
171.�Li L, Jiang H, Qiu Y, Ching WK, Vassiliadis VS. Odkrycie biomarkerów metabolitów: analiza strumienia i podejście sieci reakcji-reakcji.�BMC System Biol.�2013;7(Dodatek 2):S13.�[Artykuł bezpłatny PMC][PubMed]
172.�Patel MJ, Khalaf A, Aizenstein HJ. Badanie depresji przy użyciu metod obrazowania i uczenia maszynowego.�Klinika NeuroImage�2016;10:115.�[Artykuł bezpłatny PMC][PubMed]
173.�Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H. Produkcja cytokin i odpowiedź na leczenie w ciężkim zaburzeniu depresyjnym.Neuropsychofarmakol.�2000;22(4):370.�[PubMed]
174.�Lindqvist D, Janelidze S, Erhardt S, Trskman-Bendz L, Engstr.m G, Brundin L. Biomarkery płynu mózgowo-rdzeniowego w próbach samobójczych – analiza głównych składowych.Acta Psychiatr Scand.�2011;124(1):52.�[PubMed]
175.�Hidalgo-Mazzei D, Murru A, Reinares M, Vieta E, Colom F. Big data w zdrowiu psychicznym: trudna, fragmentaryczna przyszłość.Psychiatria Świata.�2016;15(2):186.�[Artykuł bezpłatny PMC][PubMed]
176.�Konsorcjum C-DGotPG Identyfikacja loci ryzyka o wspólnym wpływie na pięć głównych zaburzeń psychicznych: analiza całego genomu.Lancet.�2013;381(9875):1371.�[Artykuł bezpłatny PMC][PubMed]
177.�Dipnall JF, Pasco JA, Berk M i in. Łączenie eksploracji danych, uczenia maszynowego i tradycyjnych statystyk w celu wykrywania biomarkerów związanych z depresją.�PLoS Jeden.�2016;11(2):e0148195.�[Artykuł bezpłatny PMC][PubMed]
178.�K.hler O, Benros ME, Nordentoft M, et al. Wpływ leczenia przeciwzapalnego na depresję, objawy depresji i działania niepożądane: przegląd systematyczny i metaanaliza randomizowanych badań klinicznych.JAMA Psychiatria.�2014;71(12):1381.�[PubMed]
179.�Wolkowitz OM, Reus VI, Chan T, et al. Antyglukokortykoidowe leczenie depresji: podwójnie ślepa próba ketokonazolu.�Biol Psychiatria.�1999;45(8):1070.�[PubMed]
180.�McAllister-Williams RH, Anderson IM, Finkelmeyer A, et al. Wzmocnienie leków przeciwdepresyjnych metyraponem w leczeniu depresji opornej na leczenie (badanie ADD): podwójnie ślepa, randomizowana, kontrolowana placebo próba.Psychiatria Lancet.�2016;3(2):117.�[PubMed]
182.�Otte C, Hinkelmann K, Moritz S, et al. Modulacja receptora mineralokortykoidowego jako leczenie uzupełniające w depresji: randomizowane, podwójnie zaślepione, kontrolowane placebo badanie potwierdzające słuszność koncepcji.J Psychiatr Res.�2010;44(6):339.�[PubMed]
183.�Ozbolt LB, Nemeroff CB. Modulacja osi HPA w leczeniu zaburzeń nastroju.�Zaburzenia psychiatryczne.�2013;51: 1147 1154.
184.�Walker AK, Budac DP, Bisulco S i in. Blokada receptora NMDA przez ketaminę znosi wywołane przez lipopolisacharydy zachowanie podobne do depresji u myszy C57BL/6J.Neuropsychofarmakol.�2013;38(9):1609.�[Artykuł bezpłatny PMC][PubMed]
185.�Lesp�rance F, Frasure-Smith N, St-Andr� E, Turecki G, Lesp�rance P, Wiśniewski SR. Skuteczność suplementacji omega-3 w przypadku ciężkiej depresji: randomizowane, kontrolowane badanie.�J Clin Psychiatria.�2010;72(8):1054.�[PubMed]
186.�Kim S, Bae K, Kim J, et al. Stosowanie statyn w leczeniu depresji u pacjentów z ostrym zespołem wieńcowym.�Transl Psychiatria.�2015;5(8):e620.�[Artykuł bezpłatny PMC][PubMed]
187.�Shishehbor MH, Brennan ML, Aviles RJ i in. Statyny promują silne ogólnoustrojowe działanie przeciwutleniające poprzez określone szlaki zapalne.�Cyrkulacja.�2003;108(4):426.�[PubMed]
188.�Mercier A, Auger-Aubin I, Lebeau JP, et al. Dowody na przepisywanie leków przeciwdepresyjnych na schorzenia niepsychiatryczne w podstawowej opiece zdrowotnej: analiza wytycznych i przeglądy systematyczne.�Praktyka rodzinna BMC.�2013;14(1):55.�[Artykuł bezpłatny PMC][PubMed]
189.�Freland L, Beaulieu JM. Hamowanie GSK3 przez lit, od pojedynczych cząsteczek po sieci sygnalizacyjne.�Przedni Mol Neurosci.�2012;5:14.�[Artykuł bezpłatny PMC][PubMed]
190.�mgr Horowitz, Zunszain PA. Nieprawidłowości neuroimmunologiczne i neuroendokrynne w depresji: dwie strony tej samej monety.�Ann NY Acad Sci.�2015;1351(1):68.�[PubMed]
191.�Juruena MF, Cleare AJ. Nakładają się na siebie depresja atypowa, sezonowe zaburzenie afektywne i zespół przewlekłego zmęczenia.�Rev Bras Psiquiatr.�2007;29:S19�S26.�[PubMed]
192.�Castr�n E, Kojima M. Neurotroficzny czynnik pochodzenia mózgowego w zaburzeniach nastroju i leczeniu przeciwdepresyjnym.Neurobiol Dis.�2017;97(Pt B): 119.�[PubMed]
193.�Pan A, Keum N, Okereke OI, et al. Dwukierunkowy związek między depresją a zespołem metabolicznym – przegląd systematyczny i metaanaliza badań epidemiologicznych.�Opieka cukrzycowa.�2012;35(5):1171.�[Artykuł bezpłatny PMC][PubMed]
194.�Carvalho AF, Rocha DQ, McIntyre RS, et al. Adipokiny jako pojawiające się biomarkery depresji: przegląd systematyczny i metaanaliza.�J Psychiatric Res.�2014;59:28.�[PubMed]
195.�Wise T, Cleare AJ, Herane A, Young AH, Arnone D. Diagnostyczna i terapeutyczna użyteczność neuroobrazowania w depresji: przegląd.�Leczenie neuropsychiatryczne.�2014;10: 1509 1522.[Artykuł bezpłatny PMC][PubMed]
197.�Yoshimura R, Nakamura J, Shinkai K, Ueda N. Odpowiedź kliniczna na leczenie przeciwdepresyjne i poziomy 3-metoksy-4-hydroksyfenyloglikolu: mini recenzja.Prog Neuropsychopharmacol Biol Psychiatry.�2004;28(4):611.�[PubMed]
198.�Pierscionek T, Adekunte O, Watson S, Ferrier N, Alabi A. Rola kortykosteroidów w odpowiedzi przeciwdepresyjnej.ChronoPhys Ther.�2014;4: 87 98.
199.�Hage MP, Azar ST. Związek między czynnością tarczycy a depresją.�J Tarczyca Res.�2012;2012:590648.�[Artykuł bezpłatny PMC][PubMed]
200.�Dunn EC, Brown RC, Dai Y, et al. Genetyczne determinanty depresji: ostatnie odkrycia i przyszłe kierunki.�Harv Rev Psychiatria.�2015;23(1):1.�[Artykuł bezpłatny PMC][PubMed]
201.�Yang CC, Hsu YL. Przegląd opartych na akcelerometrii noszonych na ciele detektorów ruchu do monitorowania aktywności fizycznej.�Czujniki.�2010;10(8):7772.�[Artykuł bezpłatny PMC][PubMed]
Na artretyzm ból jest złożonym zjawiskiem obejmującym złożone przetwarzanie neurofizjologiczne na wszystkich poziomach szlaku bólowego. Dostępne opcje leczenia w celu złagodzenia bólu stawów są dość ograniczone, a większość pacjentów z zapaleniem stawów zgłasza jedynie niewielką ulgę w bólu przy obecnych metodach leczenia. Lepsze zrozumienie mechanizmów neuronalnych odpowiedzialnych za ból mięśniowo-szkieletowy oraz identyfikacja nowych celów pomoże w opracowaniu przyszłych terapii farmakologicznych. W tym artykule dokonano przeglądu niektórych najnowszych badań nad czynnikami, które przyczyniają się do bólu stawów i obejmują takie obszary, jak kannabinoidy, receptory aktywowane proteinazą, kanały sodowe, cytokiny i przejściowe potencjalne kanały receptorowe. Dyskutowana jest również pojawiająca się hipoteza, że choroba zwyrodnieniowa stawów może mieć komponent neuropatyczny.
Wprowadzenie
Światowa organizacja zdrowia zalicza zaburzenia mięśniowo-szkieletowe do najczęstszych przyczyn niepełnosprawności we współczesnym świecie, dotykając jedną trzecią dorosłych [1]. Jeszcze bardziej niepokojące jest to, że częstość występowania tych chorób wzrasta, podczas gdy nasza wiedza o ich przyczynach jest dość szczątkowa.
Ryc. 1 Schemat ilustrujący niektóre cele znane z modulowania bólu stawów. Neuromodulatory mogą być uwalniane z zakończeń nerwowych, a także komórek tucznych i makrofagów, aby zmienić mechanowrażliwość doprowadzającą. Endowanilloidy, kwas i szkodliwe ciepło mogą aktywować kanały jonowe waniloidalnego potencjału receptora typu 1 (TRPV1), prowadząc do uwolnienia substancji algogennej P (SP), która następnie wiąże się z receptorami neurokininy-1 (NK1). Proteazy mogą rozszczepiać i stymulować receptory aktywowane przez proteazy (PAR). Jak dotąd wykazano, że PAR2 i PAR4 uwrażliwiają pierwotne aferenty stawów. Anandamid endokannabinoidowy (AE) jest produkowany na żądanie i syntetyzowany z N-arachidonoilofosfatydyloetanoloaminy (NAPE) pod wpływem enzymatycznego działania fosfolipaz. Część AE wiąże się następnie z receptorami kannabinoidowymi-1 (CB1), prowadząc do odczulania neuronów. Niezwiązany AE jest szybko wychwytywany przez anandamidowy transporter błonowy (AMT), zanim zostanie rozłożony przez hydrolazę amidu kwasu tłuszczowego (FAAH) na etanoloaminę (Et) i kwas arachidonowy (AA). Cytokiny, czynnik martwicy nowotworu-a (TNF-a), interleukina-6 (IL-6) i interleukina1-beta (IL-1a) mogą wiązać się ze swoimi odpowiednimi receptorami, aby wzmocnić przenoszenie bólu. Wreszcie, kanały sodowe oporne na tetrodotoksynę (TTX) (Nav1.8) biorą udział w uczuleniu neuronów.
Pacjenci tęsknią za swoimi przewlekły ból zniknąć; jednak obecnie przepisywane leki przeciwbólowe są w dużej mierze nieskuteczne i towarzyszy im szereg niepożądanych skutków ubocznych. W związku z tym miliony ludzi na całym świecie cierpią z powodu wyniszczających skutków bólu stawów, na który nie ma zadowalającego leczenia [2].
Choroba zwyrodnieniowa stawów (OA) jest najczęstsza w ponad 100 różnych postaciach zapalenia stawów. OA jest postępującą chorobą zwyrodnieniową stawów, która powoduje przewlekły ból i utratę funkcji. Powszechnie OA to niezdolność stawu do skutecznej naprawy uszkodzeń w odpowiedzi na przyłożone do niego nadmierne siły. Czynniki biologiczne i psychospołeczne, które składają się na przewlekły ból w OA, nie są dobrze poznane, chociaż trwające badania odkrywają złożoną naturę objawów choroby [2]. Obecne terapie, takie jak niesteroidowe leki przeciwzapalne (NLPZ), zapewniają pewną ulgę w objawach, zmniejszając ból na krótki czas, ale nie łagodzą bólu przez całe życie pacjenta. Ponadto NLPZ w dużych dawkach nie mogą być przyjmowane wielokrotnie przez wiele lat, ponieważ może to prowadzić do toksycznego działania na nerki i krwawienia z przewodu pokarmowego.
Tradycyjnie badania nad zapaleniem stawów skupiały się głównie na chrząstce stawowej jako głównym celu terapeutycznym opracowywania nowych leków na OA do modyfikacji choroby. To ukierunkowanie na chondrogeny rzuciło nowe światło na złożone czynniki biochemiczne i biomechaniczne, które wpływają na zachowanie chondrocytów w chorych stawach. Jednakże, ponieważ chrząstka stawowa jest ukrwiona i nieunaczyniona, jest mało prawdopodobne, aby ta tkanka była źródłem bólu w OA. Ten fakt, w połączeniu z odkryciami, że nie ma korelacji między uszkodzeniem chrząstki stawowej a bólem u pacjentów z chorobą zwyrodnieniową stawów [3,4] lub przedklinicznymi modelami choroby zwyrodnieniowej stawów [5], spowodował przesunięcie uwagi na opracowywanie leków do skutecznej kontroli bólu . W tym artykule dokonamy przeglądu najnowszych wyników badań nad bólem stawów i podkreślimy niektóre z pojawiających się celów, które mogą być przyszłością leczenia bólu stawów (podsumowane na ryc. 1)
Cytokiny
Działania różnych cytokin w badaniach neurofizjologii stawów zajmują ostatnio dość znaczące miejsce. Interleukina-6 (IL-6), na przykład, jest cytokiną, która zazwyczaj wiąże się z receptorem IL-6 związanym z błoną (IL-6R). IL-6 może również sygnalizować poprzez wiązanie się z rozpuszczalnym IL-6R (SIL-6R) w celu wytworzenia kompleksu IL-6/sIL-6R. Ten kompleks IL-6/sIL-6R następnie wiąże się z podjednostką 130 (gp130) transbłonowej glikoproteiny, umożliwiając w ten sposób IL-6 sygnalizację w komórkach, które nie wykazują konstytutywnej ekspresji IL-6R związanego z błoną [25,26, XNUMX]. IL-6 i SIL-6R odgrywają kluczową rolę w ogólnoustrojowym zapaleniu i zapaleniu stawów, ponieważ w surowicy i płynie maziowym pacjentów z RZS stwierdzono podwyższenie poziomu ich obu. [27,29]. Ostatnio Vazquez i wsp. zaobserwowali, że jednoczesne podawanie IL-6/sIL-6R do kolan szczurów powodowało ból wywołany stanem zapalnym, co objawia się wzrostem odpowiedzi neuronów rdzeniowego rogu grzbietowego na mechaniczną stymulację kolana i innych części kończyny tylnej [30]. Nadpobudliwość neuronów rdzeniowych była również obserwowana, gdy IL-6/sIL-6R był nakładany lokalnie na rdzeń kręgowy. Dordzeniowe podanie rozpuszczalnego gp130 (który usuwałby kompleksy IL-6/sIL-6R, zmniejszając w ten sposób trans-sygnalizację) hamował uczulenie ośrodkowe indukowane przez IL-6/sIL-6R. Jednak doraźne podanie samego rozpuszczalnego gp130 nie zmniejszyło odpowiedzi neuronalnych na już rozwinięte zapalenie stawów.
Kanały przejściowego potencjału receptorowego (TRP) to nieselektywne kanały kationowe, które działają jako integratory różnych procesów fizjologicznych i patofizjologicznych. Oprócz termosensacji, chemosensacji i mechanosensation, kanały TRP biorą udział w modulowaniu bólu i stanu zapalnego. Na przykład wykazano, że kanały jonowe TRP waniloid-1 (TRPV1) przyczyniają się do bólu zapalnego stawów, ponieważ przeczulica cieplna nie była wywoływana u myszy z mono artretyzmem TRPV1 [31]. Podobnie, kanały jonowe TRP ankyrin-1 (TRPA1) są zaangażowane w artretyczną nadwrażliwość mechaniczną, ponieważ blokada receptora za pomocą selektywnych antagonistów łagodzi ból mechaniczny w zapaleniu modelu z kompletnym adiuwantem Freunda [32,33, 1]. Dalsze dowody na udział TRPV1 w neuroprzekaźnictwie bólu w OA pochodzą z badań, w których ekspresja neuronalna TRPV34 jest podwyższona w modelu monojodooctanu sodu w OA [1]. Dodatkowo, ogólnoustrojowe podawanie antagonisty TRPV889425 A-35 zmniejszyło wywoływaną i spontaniczną aktywność neuronów o szerokim zakresie dynamicznym w całym rdzeniu kręgowym i swoistych dla nocycepcji w modelu monojodooctanu [XNUMX]. Dane te sugerują, że endowanilloidy mogą być zaangażowane w ośrodkowe procesy sensytyzacji związane z bólem w OA.
Obecnie wiadomo, że w genie kodującym TRPV1 występują co najmniej cztery polimorfizmy prowadzące do zmiany struktury kanału jonowego i upośledzenia funkcji. Jeden szczególny polimorfizm (rs8065080) zmienia wrażliwość TRPV1 na kapsaicynę, a osoby niosące ten polimorfizm są mniej wrażliwe na przeczulicę termiczną [36]. W niedawnym badaniu zbadano, czy pacjenci z chorobą zwyrodnieniową stawów z polimorfizmem rs8065080 doświadczyli zmienionej percepcji bólu w oparciu o tę anomalię genetyczną. Zespół badawczy odkrył, że pacjenci z bezobjawową chorobą zwyrodnieniową stawu kolanowego częściej byli nosicielami genu rs8065080 niż pacjenci z bolesnymi stawami [37]. Ta obserwacja wskazuje, że pacjenci z OA funkcjonujący normalnie; Kanały TRPV1 mają zwiększone ryzyko bólu stawów i ponownie potwierdzają potencjalny udział TRPV1 w percepcji bólu w OA.
Wnioski
Podczas gdy przeszkoda w skutecznym leczeniu bólu stawów nadal pozostaje, dokonuje się ogromny postęp w naszym zrozumieniu procesów neurofizjologicznych odpowiedzialnych za powstawanie bólu stawów. Nieustannie odkrywane są nowe cele, podczas gdy mechanizmy stojące za znanymi szlakami są dalej definiowane i udoskonalane. Celowanie w jeden konkretny receptor lub kanał jonowy raczej nie będzie rozwiązaniem normalizacji bólu stawów, ale raczej wskazane jest podejście polipragmatyczne, w którym różne mediatory są stosowane w połączeniu w określonych fazach choroby. Rozwikłanie funkcjonalnych obwodów na każdym poziomie ścieżki bólu poprawi również naszą wiedzę na temat powstawania bólu stawów. Na przykład identyfikacja obwodowych mediatorów bólu stawów pozwoli nam kontrolować nocycepcję w obrębie stawu i prawdopodobnie uniknąć ośrodkowych skutków ubocznych podawanych ogólnoustrojowo leków farmakoterapeutycznych.
BÓL FACETOGENNY
ZESPÓŁ FACETA I BÓL FACETOGENICZNY
Zespół twarzy to choroba stawowa związana z lędźwiowymi stawami międzywyrostkowymi i ich unerwieniem, która powoduje zarówno miejscowy, jak i promieniujący ból facetogenny.
Nadmierna rotacja, wyprost lub zgięcie kręgosłupa (wielokrotne nadużywanie) może spowodować zmiany zwyrodnieniowe chrząstki stawu. Ponadto może obejmować zmiany zwyrodnieniowe innych struktur, w tym krążka międzykręgowego.
ZESPÓŁ TWARZY SZYJNEJ I BÓL FACETOGENICZNY
Ból osiowy szyi (rzadko promieniujący poza barki), najczęściej jednostronny.
Ból i / lub ograniczenie wydłużenia i obrotu
Czułość po palpacji
Promieniowanie bólu twarzogennego lokalnie lub na barkach lub w górnej części pleców i rzadko promieniuje z przodu lub w dół ramienia lub w palce, tak jak przepuklina dysku może.
ZESPÓŁ LĘDŹWIOWO-TWARZOWA I BÓL FACETOGENNY
Ból lub tkliwość w dolnej części pleców.
Lokalna czułość / sztywność wzdłuż kręgosłupa w dolnej części pleców.
Ból, sztywność lub trudności z niektórymi ruchami (takie jak wstawanie prosto lub wstawanie z krzesła.
Ból po przeprostaniu
Przenoszony ból z górnych stawów lędźwiowych może rozciągać się na bok, biodro i górną część bocznego uda.
Przenoszony ból z dolnych stawów kręgosłupa lędźwiowego może wnikać głęboko w udo, z boku i/lub z tyłu.
Stawy międzykręgowe L4-L5 i L5-S1 mogą odnosić się do bólu rozciągającego się do dalszej bocznej nogi, a w rzadkich przypadkach do stopy
MEDYCYNA OPARTA NA DOWODACH
Oparta na dowodach interwencyjna medycyna bólu według diagnozy klinicznej
12. Ból pochodzący ze stawów lędźwiowych
Abstrakcyjny
Chociaż istnienie zespołu fasetowego było od dawna kwestionowane, obecnie jest on powszechnie akceptowany jako jednostka kliniczna. W zależności od kryteriów diagnostycznych, stawy zygapofialne stanowią od 5% do 15% przypadków przewlekłego, osiowego bólu krzyża. Najczęściej ból facetogenny jest wynikiem powtarzającego się stresu i/lub skumulowanego urazu niskiego poziomu, prowadzącego do zapalenia i rozciągnięcia torebki stawowej. Najczęstszą dolegliwością jest osiowy ból dolnej części pleców z bólem rzutowanym odczuwanym w okolicy lędźwiowej, biodrowej i udowej. Żadne wyniki badania fizykalnego nie są patognomoniczne dla diagnozy. Najsilniejszym wskaźnikiem bólu facetogennego odcinka lędźwiowego jest zmniejszenie bólu po znieczuleniu po znieczuleniu gałęzi przyśrodkowych (gałęzi przyśrodkowych) gałęzi grzbietowych, które unerwiają stawy międzykręgowe. Ponieważ mogą wystąpić wyniki fałszywie dodatnie i ewentualnie fałszywie ujemne, wyniki należy interpretować ostrożnie. U pacjentów z potwierdzonym iniekcyjnie bólem przynasadowym stawów, interwencje proceduralne mogą być podejmowane w kontekście wielodyscyplinarnego, multimodalnego schematu leczenia, który obejmuje farmakoterapię, fizjoterapię i regularne ćwiczenia oraz, jeśli jest to wskazane, psychoterapię. Obecnie złotym standardem leczenia bólu facetogennego jest leczenie falami radiowymi (1 B+). Dowody na poparcie dostawowych kortykosteroidów są ograniczone; dlatego powinno to być zarezerwowane dla tych, którzy nie reagują na leczenie falami radiowymi (2 B1).
Ból Facetogenny emanujący ze stawów kręgosłupa lędźwiowego jest częstą przyczyną bólu krzyża w populacji dorosłych. Goldthwaite jako pierwszy opisał ten syndrom w 1911 roku, a Ghormleyowi przypisuje się ogólnie określenie „zespół fasetowy” w 1933 roku. Ból fasetogenny definiuje się jako ból, który powstaje z dowolnej struktury będącej częścią stawów fasetowych, w tym torebki włóknistej. , błony maziowej, chrząstki szklistej i kości.35
Częściej jest to wynik powtarzającego się stresu i/lub skumulowanej traumy niskiego poziomu. Prowadzi to do stanu zapalnego, który może powodować wypełnienie płynem i obrzękiem stawu międzykręgowego, co prowadzi do rozciągania torebki stawowej i późniejszego powstawania bólu.27 Zmiany zapalne wokół stawu zewnętrznego mogą również podrażniać nerw rdzeniowy przez zwężenie przedsionka, powodując rwę kulszową. Ponadto, Igarashi i wsp. 28 stwierdzili, że cytokiny zapalne uwalniane przez torebkę stawową brzuszną u pacjentów ze zwyrodnieniem stawu zygapofizycznego mogą być częściowo odpowiedzialne za objawy neuropatii u osób ze zwężeniem kręgosłupa. Czynniki predysponujące do zygapofizjologicznego bólu stawów obejmują kręgozmyk / lizę, chorobę zwyrodnieniową dysku i zaawansowany wiek.5
TESTY DODATKOWE IC
Częstość występowania zmian patologicznych w stawach międzywyrostkowych w badaniu radiologicznym zależy od średniego wieku badanych, zastosowanej techniki radiologicznej oraz definicji nieprawidłowości. Degeneracyjne stawy można najlepiej zwizualizować za pomocą tomografii komputerowej (CT).49
BÓL NEUROPATYCZNY
Ból wywołany lub spowodowany pierwotną zmianą lub dysfunkcją w somatosensorycznym układzie nerwowym.
Ból neuropatyczny jest zwykle przewlekła, trudna do leczenia i często oporna na standardowe leczenie przeciwbólowe.
Abstrakcyjny
Ból neuropatyczny jest spowodowany uszkodzeniem lub chorobą układu somatosensorycznego, w tym włókien obwodowych (włókna A?, A? i C) oraz neuronów ośrodkowych i dotyka 7-10% populacji ogólnej. Opisano wiele przyczyn bólu neuropatycznego. Częstość jego występowania prawdopodobnie wzrośnie z powodu starzenia się globalnej populacji, zwiększonej cukrzycy i poprawy przeżywalności z powodu raka po chemioterapii. Rzeczywiście, brak równowagi między pobudzającą i hamującą sygnalizacją somatosensoryczną, zmiany w kanałach jonowych i zmienność w sposobie modulowania komunikatów bólowych w ośrodkowym układzie nerwowym są powiązane z bólem neuropatycznym. Ponadto ciężar przewlekłego bólu neuropatycznego wydaje się być związany ze złożonością objawów neuropatycznych, złymi rokowaniami i trudnymi decyzjami terapeutycznymi. Co ważne, jakość życia jest obniżona u pacjentów z bólem neuropatycznym z powodu częstszych przepisywania leków i wizyt u pracowników służby zdrowia oraz zachorowalności z powodu samego bólu i wywołującej choroby. Pomimo wyzwań postęp w zrozumieniu patofizjologii bólu neuropatycznego jest bodźcem do opracowywania nowych procedur diagnostycznych i spersonalizowanych interwencji, które podkreślają potrzebę wielodyscyplinarnego podejścia do leczenia bólu neuropatycznego.
PATOGENEZA BÓLU NEUROPATYCZNEGO
MECHANIZMY PERYFERYJNE
Po uszkodzeniu nerwów obwodowych neurony stają się bardziej wrażliwe i rozwijają nieprawidłową pobudliwość i zwiększoną wrażliwość na stymulację.
Jest to znane jako ... Uczulenie peryferyjne!
MECHANIZMY CENTRALNE
W wyniku trwającej spontanicznej aktywności na obwodzie neurony rozwijają zwiększoną aktywność tła, powiększone pola recepcyjne i zwiększoną reakcję na impulsy aferentne, w tym normalne bodźce dotykowe. Jest to znane jako ... Centralne uczulenie!
Przewlekły ból neuropatyczny występuje częściej u kobiet (8% w porównaniu z 5.7% u mężczyzn) oraz u pacjentów w wieku >50 lat (8.9% w porównaniu z 5.6% w wieku <49 lat) i najczęściej dotyczy dolnej części pleców i kończyn dolnych , szyja i kończyny górne24. Bolesne radikulopatie odcinka lędźwiowego i szyjnego są prawdopodobnie najczęstszą przyczyną przewlekłego bólu neuropatycznego. Zgodnie z tymi danymi, badanie przeprowadzone wśród >12,000 40 pacjentów z przewlekłym bólem zarówno z bólem nocyceptywnym, jak i neuropatycznym, skierowane do specjalistów od bólu w Niemczech, wykazało, że 25% wszystkich pacjentów doświadczyło przynajmniej niektórych cech bólu neuropatycznego (takich jak uczucie pieczenia, drętwienie i mrowienie); Szczególnie dotknięci byli pacjenci z przewlekłym bólem pleców i radikulopatiąXNUMX.
Wkład neurofizjologii klinicznej w zrozumienie mechanizmów bólów głowy typu napięciowego.
Abstrakcyjny
Do tej pory kliniczne badania neurofizjologiczne nad napięciowymi bólami głowy (TTH) były prowadzone w dwóch głównych celach: (1) ustalenie, czy niektóre parametry neurofizjologiczne mogą działać jako markery TTH, oraz (2) zbadanie fizjopatologii TTH. Jeśli chodzi o pierwszy punkt, obecne wyniki są rozczarowujące, ponieważ pewne nieprawidłowości stwierdzone u pacjentów z TTH można często zaobserwować również u osób z migreną. Z drugiej strony neurofizjologia kliniczna odegrała ważną rolę w debacie na temat patogenezy TTH. Badania nad eksteroceptywną supresją skurczu mięśnia skroniowego wykazały zaburzenia pobudliwości pnia mózgu i kontroli suprasegmentalnej. Podobny wniosek wysunięto na podstawie odruchów trójdzielno-szyjkowych, których nieprawidłowości w TTH sugerowały zmniejszoną aktywność hamującą interneuronów pnia mózgu, co odzwierciedla nieprawidłowe endogenne mechanizmy kontroli bólu. Co ciekawe, nieprawidłowości pobudliwości nerwowej w TTH wydają się być zjawiskiem uogólnionym, nie ograniczonym do okolic czaszki. Wadliwe mechanizmy podobne do DNIC zostały rzeczywiście wykazane również w rejonach somatycznych przez badania odruchów zgięciowych nocyceptywnych. Niestety większość badań neurofizjologicznych nad TTH jest naznaczona poważnymi wadami metodologicznymi, których należy unikać w przyszłych badaniach, aby lepiej wyjaśnić mechanizmy TTH.
Referencje:
Neurofizjologia bólu stawów. McDougall JJ1 Lintona P.
Ból pochodzący ze stawów na odcinku lędźwiowym. van Kleef M1,Vanelderen P,Cohen SP,Lataster A,Van Zundert J,Mekhail N.
Ból neuropatycznyLuana Colloca,1Taylor Ludman,1Didier Bouhassira,2Ralf Baron,3Anthony H. Dickenson,4David Yarnitsky,5Roy Freeman,6Andrea Truini,7Nadine Attal, Nanna B. Finnerup,9Christopher Eccleston,10,11Eija Kalso,12David L. Bennett,13Robert H. Dworkin,14i Srinivasa N. Raja15
Wkład neurofizjologii klinicznej w zrozumienie mechanizmów bólów głowy typu napięciowego. Rossi P1, Vollono C, Valeriani M, Sandrini G.
Lekarze definiują chroniczny ból, jak każdy ból, który trwa przez 3 do 6 miesięcy lub dłużej. The ból wpływa na zdrowie psychiczne jednostki i na codzienne życie. Ból pochodzi z serii wiadomości, które przebiegają przez układ nerwowy. Depresja wydaje się podążać za bólem. Powoduje poważne objawy, które wpływają na to, jak jednostka czuje, myśli i jak radzi sobie z codziennymi czynnościami, tj. Spaniem, jedzeniem i pracą. Kręgarze, dr Alex Jimenez zgłębia potencjalne biomarkery, które mogą pomóc w znalezieniu i leczeniu przyczyn bólu i chronicznego bólu.
Pierwszym krokiem w skutecznym leczeniu bólu jest kompleksowa ocena biopsychospołeczna.
Stopień patologii organicznej może nie być dokładnie odzwierciedlony w doświadczeniu bólu.
Wstępna ocena może zostać wykorzystana do zidentyfikowania obszarów wymagających bardziej dogłębnej oceny.
Dostępnych jest wiele sprawdzonych narzędzi do samodzielnego raportowania w celu oceny wpływu chronicznego bólu.
Ocena pacjentów z przewlekłym bólem
Przewlekły ból jest problemem zdrowia publicznego dotykającym 20% populacji krajów zachodnich. Pomimo wielu postępów naukowych w zakresie rozumienia neurofizjologii bólu, precyzyjne ocenianie i diagnozowanie problemu przewlekłego bólu pacjenta nie jest proste ani dobrze zdefiniowane. W jaki sposób chroniczny ból jest konceptualizowany, wpływa na ocenę bólu i czynników uwzględnianych podczas diagnozy bólu przewlekłego. Nie ma relacji jeden do jednego między ilością lub rodzajem patologii organicznej a natężeniem bólu, ale zamiast tego doświadczenie bólu przewlekłego kształtowane jest przez niezliczoną liczbę biomedycznych, psychospołecznych (np. wierzenia pacjentów, oczekiwania i nastrój) oraz czynników behawioralnych (np. kontekst, reakcje znaczących innych). Ocena każdej z tych trzech dziedzin poprzez kompleksową ocenę osoby z przewlekłym bólem jest niezbędna do podjęcia decyzji o leczeniu i uzyskania optymalnych wyników. Ocena ta powinna obejmować dokładną historię pacjenta i ocenę lekarską oraz krótką rozmowę kwalifikacyjną, podczas której można zaobserwować zachowanie pacjenta. Dalsza ocena w celu odpowiedzi na pytania postawione podczas wstępnej oceny będzie decydować o tym, jakie dodatkowe oceny, jeśli są, mogą być odpowiednie. Dostępne są standaryzowane, samodzielnie zgłaszane instrumenty do oceny natężenia bólu pacjenta, zdolności funkcjonalnych, przekonań i oczekiwań oraz emocjonalnego dystresu, którymi może zarządzać lekarz, lub można skierować do dogłębnej oceny, aby pomóc w planowaniu leczenia.
Ból jest niezwykle powszechnym objawem. Szacuje się, że sam przewlekły ból wpływa na 30% dorosłej populacji USA, powyżej 100 milionów dorosłych.1
Pomimo rosnących kosztów leczenia osób z przewlekłym bólem, ulga dla wielu pozostaje nieuchwytna i całkowite wyeliminowanie bólu jest rzadkie. Chociaż nastąpił znaczny postęp w wiedzy z zakresu neurofizjologii bólu, wraz z rozwojem silnych leków przeciwbólowych i innych innowacyjnych interwencji medycznych i chirurgicznych, średnia redukcja bólu dzięki dostępnym procedurom wynosi 30% i ma to miejsce w mniej niż połowa leczonych pacjentów.
Sposób, w jaki myślimy o bólu, wpływa na sposób, w jaki oceniamy ból. Ocena rozpoczyna się od wywiadu i badania fizykalnego, a następnie testów laboratoryjnych i diagnostycznych procedur obrazowania w celu zidentyfikowania i / lub potwierdzenia obecności jakiejkolwiek podstawowej patologii powodującej objawy lub objawy. generator bólu.
W przypadku braku możliwej do zidentyfikowania patologii organicznej, podmiot świadczący opiekę zdrowotną może założyć, że zgłoszenie objawów wynika z czynników psychologicznych i może poprosić o ocenę psychologiczną w celu wykrycia czynników emocjonalnych leżących u podstaw raportu pacjenta. Istnieje dwoistość, w której raport o objawach przypisywany jest któremuś z nich somatyczny or mechanizmy psychogenne.
Jako przykład można podać organiczne zasady niektórych najczęściej występujących i nawracających ostrych (np. Ból głowy) 3 i przewlekłych [np. ból pleców, problemy z bólem fibromialgii (FM)] są w dużej mierze nieznane, 4,5, natomiast osoby bezobjawowe mogą mieć nieprawidłowości strukturalne, takie jak przepuklina krążków, które wyjaśniałyby ból, gdyby był obecny. 6,7Brakuje adekwatnych wyjaśnień dla pacjentów bez zidentyfikowanej patologii organicznej, którzy zgłaszają silny ból i osoby wolne od bólu z istotną, obiektywną patologią.
Ból przewlekły dotyka nie tylko indywidualnego pacjenta, ale także jego znaczących innych (partnerów, krewnych, pracodawców, współpracowników i przyjaciół), niezbędne jest odpowiednie leczenie. Zadowalające leczenie może pochodzić wyłącznie z kompleksowej oceny biologicznej etiologii bólu w połączeniu ze specyficzną prezentacją psychospołeczną i behawioralną pacjenta, w tym z jego stanem emocjonalnym (np. Lęk, depresja i gniew), percepcją i zrozumieniem objawów oraz reakcjami na te objawy znaczących innych.8,9 Głównym założeniem jest, że wiele czynników wpływa na objawy i ograniczenia funkcjonalne osób z przewlekłym bólem. Dlatego potrzebna jest kompleksowa ocena, która dotyczy dziedzin biomedycznych, psychospołecznych i behawioralnych, ponieważ każdy z nich przyczynia się do chronicznego bólu i związanej z nim niepełnosprawności. 10,11
Kompleksowa ocena osoby z przewlekłym bólem
Turk i Meichenbaum12 zasugerowali, że trzy centralne pytania powinny kierować oceną osób zgłaszających ból:
Jaki jest zakres choroby lub urazu pacjenta (upośledzenie fizyczne)?
Jaka jest wielkość choroby? Oznacza to, w jakim stopniu pacjent cierpi, jest niepełnosprawny i nie może cieszyć się zwykłymi czynnościami?
Czy zachowanie jednostki wydaje się odpowiednie dla choroby lub urazu, czy też istnieją dowody na nasilenie objawów z różnych przyczyn psychologicznych lub społecznych (np. Korzyści, takie jak pozytywna uwaga, leki zmieniające nastrój, rekompensata finansowa)?
Aby odpowiedzieć na te pytania, należy zebrać informacje od pacjenta na podstawie wywiadu i badania przedmiotowego, w połączeniu z wywiadem klinicznym oraz za pomocą standardowych narzędzi oceny. Pracownicy służby zdrowia muszą poszukiwać jakiejkolwiek przyczyny (przyczyn) bólu poprzez badanie fizykalne i testy diagnostyczne, jednocześnie oceniając nastrój pacjenta, lęki, oczekiwania, wysiłki radzenia sobie, zasoby, reakcje bliskich osób oraz wpływ bólu na pacjentów mieszka 11 Krótko mówiąc, lekarz musi ocenić `` całą osobę '', a nie tylko ból.
Ogólnymi celami historii i oceny medycznej są:
(i) określają potrzebę dodatkowych badań diagnostycznych
(ii) określić, czy dane medyczne mogą wyjaśnić objawy, nasilenie objawów i ograniczenia funkcjonalne pacjenta
(iii) postawić diagnozę medyczną
(iv) ocenić dostępność odpowiedniego leczenia
(v) ustalić cele leczenia
(vi) określić właściwy przebieg zarządzania objawami, jeżeli całkowite wyleczenie nie jest możliwe.
Znaczna liczba pacjentów, którzy zgłaszają ból przewlekły, nie wykazuje żadnej fizycznej patologii za pomocą zwykłych radiogramów, obliczonych tomografii osiowej lub elektromografii (dostępna jest obszerna literatura na temat oceny fizycznej, procedur oceny radiologicznej i laboratoryjnej w celu określenia fizycznej podstawy bólu), 17 sprawia, że precyzyjna diagnostyka patologiczna jest trudna lub niemożliwa.
Pomimo tych ograniczeń historia pacjenta i badanie przedmiotowe pozostają podstawą diagnozy medycznej, mogą stanowić zabezpieczenie przed interpretacją ustaleń z obrazowania diagnostycznego, które są w dużej mierze potwierdzające i mogą być wykorzystane do ukierunkowania dalszych działań ewaluacyjnych.
Ponadto pacjenci z przewlekłymi problemami bólowymi często spożywają różnorodne leki.18 Ważne jest, aby omówić bieżące leki pacjenta podczas rozmowy kwalifikacyjnej, ponieważ wiele leków przeciwbólowych wiąże się z działaniami niepożądanymi, które mogą powodować lub imitować problemy emocjonalne. 19 Pracownicy służby zdrowia powinni nie tylko znać leki stosowane w leczeniu bólu przewlekłego, ale także skutki uboczne tych leków, które powodują zmęczenie, trudności ze snem i zmiany nastroju, aby uniknąć błędnej diagnozy depresji.
Uważa się, że codzienne dzienniki są dokładniejsze, ponieważ opierają się raczej na czasie rzeczywistym niż na przypomnieniu. Pacjenci mogą zostać poproszeni o utrzymywanie regularnych dzienników natężenia bólu z ocenami rejestrowanymi kilka razy dziennie (np. Posiłki i pora snu) przez kilka dni lub tygodni, a wielokrotne oceny bólu mogą być uśredniane w czasie.
Jednym z problemów zauważonych przy korzystaniu z dzienników papierowo-ołówkowych jest to, że pacjenci mogą nie stosować się do instrukcji, aby podawać oceny w określonych odstępach czasu. Pacjenci mogą raczej wypełniać dzienniki z wyprzedzeniem (`` wypełnij do przodu '') lub na krótko przed wizytą u lekarza (`` wypełnij wstecz ''), 24 podważając domniemaną ważność dzienników. Dzienniki elektroniczne zyskały akceptację w niektórych badaniach naukowych, aby uniknąć tych problemów.
Badania wykazały znaczenie oceny ogólnej jakości życia związanej ze zdrowiem (HRQOL) u pacjentów z przewlekłym bólem oprócz funkcji. 31,32 Istnieje wiele dobrze ustalonych, wspieranych psychometrycznie środków HRQOL [Badanie wyników krótkookresowych badań lekarskich dotyczących zdrowia (SF -36)], ogólne miary funkcjonowania fizycznego 33 [np. Wskaźnik niepełnosprawności (PDI)], 34 i środki specyficzne dla choroby [np. Zachodni Ontario MacMaster Indeks choroby zwyrodnieniowej stawów (WOMAC), 35 Roland-Morris Backability Disability Questionnaire (RDQ)] 36 do oceny funkcji i jakości życia.
Działania specyficzne dla choroby mają na celu ocenę wpływu konkretnego schorzenia (np. Bólu i sztywności u osób z chorobą zwyrodnieniową stawów), podczas gdy środki ogólne umożliwiają porównanie funkcjonowania fizycznego związanego z danym zaburzeniem i jego leczenia z różnymi innymi schorzeniami. Specyficzne skutki zaburzenia mogą nie zostać wykryte podczas korzystania z ogólnej miary; w związku z tym, środki specyficzne dla choroby mogą wykazywać klinicznie istotne polepszenie lub pogorszenie określonych funkcji w wyniku leczenia. Ogólne miary funkcjonowania mogą być przydatne do porównywania pacjentów z różnymi bolesnymi warunkami. Łączne stosowanie środków specyficznych dla choroby i generycznych ułatwia osiągnięcie obu celów.
Obecność stresu emocjonalnego u osób z przewlekłym bólem stanowi wyzwanie przy ocenie objawów takich jak zmęczenie, obniżony poziom aktywności, obniżone libido, zmiana apetytu, zaburzenia snu, przyrost masy ciała lub utrata wagi oraz zaburzenia pamięci i stężenia, ponieważ objawy te mogą być objawami w wyniku bólu, bólu emocjonalnego lub leków przepisywanych w celu opanowania bólu.
Instrumenty zostały opracowane specjalnie z myślą o pacjentach cierpiących na ból w celu oceny cierpienia psychicznego, wpływu bólu na życie pacjentów, poczucia kontroli, zachowań radzenia sobie oraz postaw wobec choroby, bólu i pracowników służby zdrowia17.
Na przykład: Inwentarz Depresji Becka (BDI) 39 i Profil Stanów Nastroju (POMS) 40 są psychometrycznie poprawne w ocenie objawów depresji, zaburzeń emocjonalnych i zaburzeń nastroju, i zaleca się ich stosowanie we wszystkich badaniach klinicznych przewlekły ból; 41 jednak, wyniki muszą być interpretowane z ostrożnością, a kryteria poziomu emocjonalnego dystresu mogą wymagać modyfikacji, aby zapobiec fałszywym pozytywnym wynikom. 42
Lab Biomarkers for Pain
Biomarkery to cechy biologiczne, które można wykorzystać do wskazania zdrowia lub choroby. W niniejszym artykule dokonano przeglądu badań dotyczących biomarkerów bólu krzyża (LBP) u ludzi. LBP jest główną przyczyną niepełnosprawności, spowodowanej różnymi zaburzeniami związanymi z kręgosłupem, w tym zwyrodnieniem krążka międzykręgowego, przepuklinami tarczy, zwężeniem kręgosłupa i artretyzmem stawów. Badania te koncentrują się na mediatorach zapalnych, ponieważ stan zapalny przyczynia się do patogenezy degeneracji dysków i powiązanych mechanizmów bólowych. Coraz częściej badania sugerują, że obecność mediatorów stanu zapalnego można mierzyć ogólnoustrojowo we krwi. Te biomarkery mogą służyć jako nowe narzędzia do kierowania opieką nad pacjentem. Obecnie odpowiedź pacjenta na leczenie jest nieprzewidywalna ze znacznym odsetkiem nawrotów, a chociaż zabiegi chirurgiczne mogą zapewniać korektę anatomiczną i łagodzenie bólu, są inwazyjne i kosztowne. Przegląd obejmuje badania przeprowadzone na populacjach o określonych diagnozach i nieokreślonym pochodzeniu LBP. Ponieważ naturalna historia LBP postępuje, czasowy charakter badań jest kategoryzowany przez czas trwania symptomologii / choroby. Powiązane badania dotyczące zmian w biomarkerach z leczeniem są również przeglądane. Ostatecznie, biomarkery diagnostyczne LBP i degeneracja kręgosłupa mają potencjał, aby zapoczątkować erę zindywidualizowanej medycyny kręgosłupa dla spersonalizowanych terapii w leczeniu LBP.
Biomarkery do przewlekłego bólu neuropatycznego i potencjalne zastosowanie w stymulacji rdzenia kręgowego
Przegląd ten skupił się na zrozumieniu, które substancje w ciele człowieka zwiększają się i zmniejszają wraz ze wzrostem bólu neuropatycznego. Przeanalizowaliśmy różne badania i zobaczyliśmy korelacje pomiędzy bólem neuropatycznym a komponentami układu odpornościowego (system ten chroni organizm przed chorobami i infekcjami). Nasze odkrycia będą szczególnie przydatne w zrozumieniu sposobów zmniejszenia lub wyeliminowania dyskomfortu, co wiąże się z przewlekłym bólem neuropatycznym. Procedura stymulacji rdzenia kręgowego (SCS) jest jedną z niewielu dość skutecznych metod leczenia bólu. Badanie uzupełniające zastosuje nasze ustalenia z tego przeglądu do SCS, w celu zrozumienia mechanizmu i dalszej optymalizacji skuteczności.
Stwierdzono, że cytokiny prozapalne, takie jak IL-1 p, IL-6, IL-2, IL-33, CCL3, CXCL1, CCR5 i TNF-a, odgrywają znaczącą rolę w amplifikacji przewlekłych stanów bólowych.
Po przeglądzie różnych badań dotyczących biomarkerów bólu stwierdziliśmy, że poziomy w surowicy cytokin prozapalnych i chemokin, takich jak IL-1 p, IL-6, IL-2, IL-33, CCL3, CXCL1, CCR5 i TNF - ?, były znacznie podwyższone podczas doświadczania bólu przewlekłego. Z drugiej strony, stwierdzono, że cytokiny przeciwzapalne, takie jak IL-10 i IL-4, wykazują znaczną regulację w dół podczas przewlekłego stanu bólowego.
Biomarkery na depresję
Ogrom badań wiązał się z setkami przypuszczalnych biomarkerów depresji, ale jeszcze nie w pełni wyjaśnił ich rolę w chorobie depresyjnej lub ustalił, co jest nieprawidłowe, w jakich przypadkach pacjenci i jak informacje biologiczne mogą być wykorzystywane do poprawy diagnozy, leczenia i prognozy. Ten brak postępu jest częściowo spowodowany naturą i heterogenicznością depresji, w połączeniu z metodologiczną heterogenicznością w literaturze badawczej i szeroką gamą biomarkerów z potencjałem, których ekspresja często zmienia się w zależności od wielu czynników. Przeglądamy dostępną literaturę, która wskazuje, że markery zaangażowane w procesy zapalne, neurotroficzne i metaboliczne, a także neuroprzekaźnik i elementy układu neuroendokrynnego, są bardzo obiecującymi kandydatami. Można je zmierzyć za pomocą oceny genetycznej i epigenetycznej, transkryptomicznej i proteomicznej, metabolomicznej i neuroobrazowej. Obecnie konieczne jest zastosowanie nowatorskich podejść i systematycznych programów badawczych w celu ustalenia, czy i które biomarkery można wykorzystać do przewidywania odpowiedzi na leczenie, stratyfikacja pacjentów do określonych terapii i opracować cele dla nowych interwencji. Doszliśmy do wniosku, że istnieje wiele obietnic redukcji obciążenia depresją poprzez dalsze rozwijanie i rozszerzanie tych możliwości badawczych.
Referencje:
Ocena pacjentów z przewlekłym bólem EJ Dansiet i DC Turk * t
Biomarkery zapalne bólów krzyża i zwyrodnienia krążków: przegląd.
Khan AN1, Jacobsen HE2, Khan J1, Filippi CG3, Levine M3, Lehman RA Jr2,4, Riew KD2,4, Lenke LG2,4, Chahine NO2,5.
Biomarkery przewlekłego bólu neuropatycznego i ich potencjalne zastosowanie w stymulacji rdzenia kręgowego: przegląd
Chibueze D. Nwagwu, 1 Christina Sarris, MD, 3 Yuan-Xiang Tao, Ph.D., MD, 2 i Antonios Mammis, MD1,2
Biomarkery na depresję: najnowsze spostrzeżenia, aktualne wyzwania i perspektywy na przyszłość. Strawbridge R1, Young AH1,2, Cleare AJ1,2.
Ból jest naturalną reakcją organizmu na uraz lub chorobę i często jest ostrzeżeniem, że coś jest nie tak. Gdy problem zostanie wyleczony, na ogół przestajemy odczuwać bolesne objawy, jednak co się dzieje, gdy ból utrzymuje się długo po ustąpieniu przyczyny? Chroniczny ból jest medycznie definiowany jako uporczywy ból, który trwa od 3 do 6 miesięcy lub dłużej. Przewlekły ból jest z pewnością stanem trudnym do życia, wpływającym na wszystko, począwszy od poziomu aktywności jednostki i jej zdolności do pracy, a także na jej osobiste relacje i stan psychiczny. Ale czy zdajesz sobie sprawę, że przewlekły ból może również wpływać na strukturę i funkcję Twojego mózgu? Okazuje się, że te zmiany w mózgu mogą prowadzić zarówno do upośledzenia funkcji poznawczych, jak i psychicznych.
Przewlekły ból nie wpływa tylko na pojedynczy obszar umysłu, w rzeczywistości może powodować zmiany w wielu istotnych obszarach mózgu, z których większość jest zaangażowana w wiele podstawowych procesów i funkcji. Różne badania naukowe na przestrzeni lat wykazały zmiany w hipokampie, wraz z redukcją istoty szarej z grzbietowo-bocznej kory przedczołowej, ciała migdałowatego, pnia mózgu i prawej kory wyspy, by wymienić tylko kilka, związanych z przewlekłym bólem. Podział kilku struktur tych regionów i związanych z nimi funkcji może pomóc umieścić te zmiany w mózgu w kontekście wielu osób z przewlekłym bólem. Celem niniejszego artykułu jest wykazanie i omówienie strukturalnych i funkcjonalnych zmian w mózgu związanych z przewlekłym bólem, szczególnie w przypadku, gdy prawdopodobnie nie odzwierciedlają one ani uszkodzeń, ani atrofii.
Zmiany strukturalne mózgu w przewlekłym bólu prawdopodobnie nie odzwierciedlają ani uszkodzeń, ani atrofii
Abstrakcyjny
Wydaje się, że przewlekły ból jest związany z redukcją istoty szarej mózgu w obszarach, które można przypisać transmisji bólu. Procesy morfologiczne leżące u podstaw tych zmian strukturalnych, prawdopodobnie po reorganizacji funkcjonalnej i plastyczności centralnej w mózgu, pozostają niejasne. Ból w chorobie zwyrodnieniowej stawu biodrowego jest jednym z nielicznych zespołów bólu przewlekłego, które są zasadniczo uleczalne. Przebadaliśmy 20 pacjentów z przewlekłym bólem spowodowanym jednostronną chorobą zwyrodnieniową stawów (średnia wieku 63.25, 9.46 (SD) lat, 10 kobiet) przed operacją endoprotezy stawu biodrowego (stan bólu) i monitorowaliśmy zmiany strukturalne mózgu do 1 roku po operacji: 6 tygodnia , 8 tygodnia i 12 miesiąca, gdy jest całkowicie bezbolesny. Pacjenci z przewlekłym bólem z powodu jednostronnej choroby zwyrodnieniowej stawów mieli znacznie mniej istoty szarej w porównaniu z grupą kontrolną w korze przedniej części zakrętu obręczy (ACC), korze wyspowej i wieczko, grzbietowo-bocznej korze przedczołowej (DLPFC) i korze oczodołowo-czołowej. Regiony te funkcjonują jako struktury multiintegracyjne podczas doświadczania i przewidywania bólu. Gdy pacjenci nie odczuwali bólu po wyzdrowieniu po operacji endoprotezy, stwierdzono wzrost istoty szarej w prawie tych samych obszarach. Odkryliśmy również postępujący wzrost istoty szarej mózgu w korze przedruchowej i dodatkowym obszarze motorycznym (SMA). Dochodzimy do wniosku, że nieprawidłowości istoty szarej w przewlekłym bólu nie są przyczyną, ale wtórne do choroby i przynajmniej częściowo wynikają ze zmian funkcji motorycznych i integracji ciała.
Wprowadzenie
Dowody na funkcjonalną i strukturalną reorganizację u pacjentów z przewlekłym bólem potwierdzają pogląd, że przewlekły ból powinien być nie tylko konceptualizowany jako zmieniony stan funkcjonalny, ale także jako konsekwencja funkcjonalnej i strukturalnej plastyczności mózgu [1], [2], [3], [4], [5], [6]. W ciągu ostatnich sześciu lat opublikowano ponad 20 badań wykazujących strukturalne zmiany w mózgu w 14 przewlekłych zespołach bólowych. Uderzającą cechą wszystkich tych badań jest fakt, że zmiany istoty szarej nie były rozłożone losowo, ale występowały w określonych i funkcjonalnie wysoce specyficznych obszarach mózgu – a mianowicie zaangażowaniu w nadrdzeniowe przetwarzanie nocyceptywne. Najbardziej widoczne wyniki były różne dla każdego zespołu bólowego, ale nakładały się w korze obręczy, korze oczodołowo-czołowej, wyspie i mostku grzbietowym [4]. Inne struktury obejmują wzgórze, grzbietowo-boczną korę przedczołową, zwoje podstawy i hipokamp. Odkrycia te są często omawiane jako atrofia komórek, wzmacniając ideę uszkodzenia lub utraty istoty szarej mózgu [7], [8], [9]. W rzeczywistości naukowcy odkryli korelację między spadkiem istoty szarej mózgu a czasem trwania bólu [6], [10]. Jednak czas trwania bólu jest również powiązany z wiekiem pacjenta, a globalny, ale także regionalnie specyficzny spadek istoty szarej, zależny od wieku, jest dobrze udokumentowany [11]. Z drugiej strony, tymi zmianami strukturalnymi może być również zmniejszenie wielkości komórek, płynów pozakomórkowych, synaptogenezy, angiogenezy, a nawet zmiany objętości krwi [4], [12], [13]. Bez względu na źródło, dla naszej interpretacji takich wyników ważne jest, aby zobaczyć te wyniki badań morfometrycznych w świetle wielu badań morfometrycznych dotyczących plastyczności zależnej od ćwiczeń, biorąc pod uwagę, że po ćwiczeniach poznawczych i fizycznych wielokrotnie wykazano regionalne zmiany strukturalne mózgu [ 14].
Nie jest zrozumiałe, dlaczego tylko stosunkowo niewielka część ludzi rozwija zespół przewlekłego bólu, biorąc pod uwagę, że ból jest doświadczeniem uniwersalnym. Powstaje pytanie, czy u niektórych ludzi strukturalna różnica w centralnych układach przenoszenia bólu może działać jako skaza na przewlekły ból. Zmiany istoty szarej w bólu fantomowym z powodu amputacji [15] i urazu rdzenia kręgowego [3] wskazują, że zmiany morfologiczne mózgu są przynajmniej częściowo konsekwencją bólu przewlekłego. Jednak ból w chorobie zwyrodnieniowej stawu biodrowego (OA) jest jednym z nielicznych zespołów bólu przewlekłego, który jest zasadniczo uleczalny, ponieważ 88% tych pacjentów regularnie nie odczuwa bólu po operacji całkowitej alloplastyki stawu biodrowego (THR) [16]. W badaniu pilotażowym przeanalizowaliśmy dziesięciu pacjentów z chorobą zwyrodnieniową stawu biodrowego przed i wkrótce po operacji. Stwierdziliśmy zmniejszenie istoty szarej w przedniej części kory obręczy (ACC) i wysepce podczas przewlekłego bólu przed operacją THR i stwierdziliśmy wzrost istoty szarej w odpowiednich obszarach mózgu w stanie bezbolesnym po operacji [17]. Koncentrując się na tym wyniku, rozszerzyliśmy teraz nasze badania, badając większą liczbę pacjentów (n?=?20) po udanej THR i monitorując strukturalne zmiany mózgu w czterech odstępach czasu, do jednego roku po operacji. Aby kontrolować zmiany istoty szarej spowodowane poprawą motoryki lub depresją, przeprowadziliśmy również kwestionariusze mające na celu poprawę funkcji motorycznych i zdrowia psychicznego.
Materiały i Metody
Wolontariusze
Opisani tu pacjenci to podgrupa 20 pacjentów z 32 opublikowanych w ostatnim czasie, których porównano ze zdrową grupą kontrolną dobraną pod względem wieku i płci [17], ale uczestniczyli w dodatkowym rocznym badaniu kontrolnym. Po zabiegu 12 pacjentów zrezygnowało z drugiego zabiegu endoprotetycznego (n==2), ciężkiej choroby (n==2) i wycofania zgody (n==8). Pozostawiło to grupę dwudziestu pacjentów z jednostronną pierwotną chorobą zwyrodnieniową stawu biodrowego (średni wiek 63.25 (SD) lat, 9.46 kobiet), którzy byli badani czterokrotnie: przed operacją (stan bólu) i ponownie 10 i 6 tygodni i 8 �12 miesięcy po operacji endoprotezy, kiedy całkowicie bezbolesne. Wszyscy pacjenci z pierwotną chorobą zwyrodnieniową stawu biodrowego mieli historię bólu dłużej niż 18 miesięcy, w zakresie od 10 do 14 lat (średnia 12 lat) i średni wynik bólu 1 (w zakresie od 33 do 7.35) w wizualnej skali analogowej (VAS) w zakresie od 65.5 (brak bólu) do 40 (najgorszy wyobrażalny ból). Oceniliśmy każde wystąpienie drobnych zdarzeń bólowych, w tym zębów, uszu i bólów głowy do 90 tygodni przed badaniem. Wybraliśmy również losowo dane z 0 osób zdrowych, dopasowanych pod względem płci i wieku (średnia wieku 100 (SD) lat, 4 kobiet) z 20 wspomnianych powyżej badań pilotażowych [60,95]. Żaden z 8,52 pacjentów ani z 10 zdrowych ochotników pasujących pod względem płci i wieku nie miał historii chorób neurologicznych lub wewnętrznych. Badanie zostało zatwierdzone przez lokalną komisję ds. etyki, a przed badaniem uzyskano pisemną świadomą zgodę od wszystkich uczestników badania.
Dane behawioralne
Zebraliśmy dane dotyczące depresji, somatyzacji, lęku, bólu oraz zdrowia fizycznego i psychicznego u wszystkich pacjentów we wszystkich czterech punktach czasowych za pomocą następujących standaryzowanych kwestionariuszy: Beck Depression Inventory (BDI) [18], Brief Symptom Inventory (BSI) [19], Schmerzempfindungs-Skala (SES?=?skala nieprzyjemności bólu) [20] i 36-elementowy formularz ankiety dotyczącej zdrowia (SF-36) [21] oraz Profil Zdrowia Nottingham (NHP). Przeprowadziliśmy ANOVA z powtarzanymi pomiarami i sparowaliśmy dwustronne testy t, aby przeanalizować podłużne dane behawioralne przy użyciu SPSS 13.0 dla Windows (SPSS Inc., Chicago, IL) i zastosowaliśmy korekcję Greenhouse Geisser, jeśli naruszono założenie sferyczności. Poziom istotności ustalono na p<0.05.
VBM – akwizycja danych
Nabycie obrazu. Skanowanie MR o wysokiej rozdzielczości przeprowadzono w systemie 3T MRI (Siemens Trio) ze standardową 12-kanałową cewką nagłowną. Dla każdego z czterech punktów czasowych skan I (od 1 dnia do 3 miesięcy przed zabiegiem endoprotezy), skan II (6 do 8 tygodni po zabiegu), skan III (12 do 18 tygodni po zabiegu) i skan IV (10 14 miesięcy po zabiegu), dla każdego pacjenta uzyskano strukturalny rezonans magnetyczny T1 ważony przy użyciu sekwencji 3D-FLASH (TR 15 ms, TE 4.9 ms, kąt odchylenia 25, 1 mm warstwy, FOV 256, rozmiar woksela 256� 1 mm).
Przetwarzanie obrazu i analiza statystyczna
Wstępne przetwarzanie i analizę danych przeprowadzono za pomocą SPM2 (Wellcome Department of Cognitive Neurology, Londyn, Wielka Brytania) działającego pod kontrolą Matlab (Mathworks, Sherborn, MA, USA) i zawierającego zestaw narzędzi do morfometrii opartej na wokselach (VBM) dla danych podłużnych, które opiera się na wysokiej rozdzielczości strukturalnych obrazach 3D MR i pozwala na zastosowanie statystyk wokselowych do wykrywania regionalnych różnic w gęstości lub objętości istoty szarej [22], [23]. Podsumowując, przetwarzanie wstępne obejmowało normalizację przestrzenną, segmentację istoty szarej i wygładzanie przestrzenne 10 mm z jądrem Gaussa. Do etapów wstępnego przetwarzania zastosowaliśmy zoptymalizowany protokół [22], [23] oraz szablon istoty szarej specyficzny dla skanera i badania [17]. Zastosowaliśmy SPM2 zamiast SPM5 lub SPM8, aby uczynić tę analizę porównywalną z naszym badaniem pilotażowym [17]. ponieważ umożliwia doskonałą normalizację i segmentację danych podłużnych. Jednak, ponieważ niedawno pojawiła się nowsza aktualizacja VBM (VBM8) (dbm.neuro.uni-jena.de/vbm/), użyliśmy również VBM8.
Analiza przekrojowa
Zastosowaliśmy dwupróbkowy test t w celu wykrycia regionalnych różnic w istocie szarej mózgu między grupami (pacjenci w punkcie czasowym I (przewlekły ból) i zdrowi kontrolni). Zastosowaliśmy próg p<0.001 (nieskorygowany) w całym mózgu ze względu na naszą silną hipotezę a priory, opartą na 9 niezależnych badaniach i kohortach wykazujących zmniejszenie istoty szarej u pacjentów z przewlekłym bólem [7], [8], [ 9], [15], [24], [25], [26], [27], [28], że wzrost istoty szarej pojawi się w tych samych (dla potrzeb przetwarzania bólu) regionach, jak w naszym badaniu pilotażowym (17 ). Grupy zostały dopasowane pod względem wieku i płci bez istotnych różnic między grupami. Aby zbadać, czy różnice między grupami zmieniły się po roku, porównaliśmy również pacjentów w punkcie czasowym IV (bezbolesny, roczny okres obserwacji) z naszą zdrową grupą kontrolną.
Analiza podłużna
W celu wykrycia różnic między punktami czasowymi (Skan I�IV) porównaliśmy skany przed operacją (stan bólu) oraz ponownie 6 i 8 tygodnia i 12 miesiąca po zabiegu endoprotezy (bez bólu) jako ANOVA z powtarzanym pomiarem. Ponieważ wszelkie zmiany w mózgu spowodowane przewlekłym bólem mogą wymagać pewnego czasu, aby cofnąć się po operacji i ustąpieniu bólu oraz z powodu bólu pooperacyjnego zgłaszanego przez pacjentów, porównaliśmy w analizie podłużnej skan I i II ze skanem III i IV. Aby wykryć zmiany, które nie są ściśle związane z bólem, szukaliśmy również postępujących zmian we wszystkich przedziałach czasowych. Przerzuciliśmy mózgi pacjentów z chorobą zwyrodnieniową lewego biodra (n?=?18) w celu normalizacji bólu po stronie zarówno dla porównania grup, jak i analizy podłużnej, ale przede wszystkim przeanalizowaliśmy dane nieodwrócone. Użyliśmy wyniku BDI jako współzmiennej w modelu.
Efekt
Dane behawioralne
Wszyscy pacjenci zgłaszali przewlekły ból biodra przed operacją i nie odczuwali bólu (w odniesieniu do tego przewlekłego bólu) bezpośrednio po operacji, ale zgłaszali raczej ostry ból pooperacyjny w skanie II, który różnił się od bólu spowodowanego chorobą zwyrodnieniową stawów. Wynik zdrowia psychicznego SF-36 (F(1.925/17.322)?=?0.352, p?=?0.7) i globalny wynik BSI GSI (F(1.706/27.302)?=?3.189, p?=?0.064 ) nie wykazały żadnych zmian w czasie ani współwystępowania chorób psychicznych. Żadna z kontroli nie zgłosiła ostrego lub przewlekłego bólu i żadna nie wykazywała żadnych objawów depresji lub niepełnosprawności fizycznej/umysłowej.
Przed operacją niektórzy pacjenci wykazywali łagodne do umiarkowanych objawy depresji w wynikach BDI, które istotnie zmniejszyły się na skanie III (t(17)?=?2.317, p?=?0.033) i IV (t(16)?=?2.132, p? =?0.049). Dodatkowo, wyniki SES (nieprzyjemność bólowa) wszystkich pacjentów uległy znacznej poprawie od I (przed operacją) do II (t(16)?=?4.676, p<0.001), III (t(14)?=?). 4.760, p<0.001 i skan IV (t(14)?=?4.981, p<0.001, 1 rok po zabiegu), ponieważ nieprzyjemność bólu zmniejszała się wraz z nasileniem bólu. Ocena bólu na skanie 1 i 2 była pozytywna, ta sama ocena w dniu 3 i 4 negatywna. SES opisuje jedynie jakość odczuwanego bólu. Wynik był zatem dodatni w dniu 1 i 2 (średnio 19.6 w dniu 1 i 13.5 w dniu 2) oraz ujemny (nie dotyczy) w dniu 3 i 4. Jednak niektórzy pacjenci nie rozumieli tej procedury i stosowali SES jako ogólną jakość miary życia. Dlatego wszyscy pacjenci byli pytani tego samego dnia indywidualnie i przez tę samą osobę o wystąpienie bólu.
W skróconej formie ankiety dotyczącej stanu zdrowia (SF-36), która składa się z sumarycznych pomiarów punktacji zdrowia fizycznego i punktacji zdrowia psychicznego [29], pacjenci znacznie poprawili wynik w zakresie zdrowia fizycznego od skanu I do skanu II (t( 17)?=??4.266, p?=?0.001), skan III (t(16)?=??8.584, p<0.001 i IV (t(12)?=??7.148, p<0.001), ale nie w Skali Zdrowia Psychicznego. Wyniki NHP były zbliżone, w podskali „ból” (odwrócona polaryzacja) zaobserwowano istotną zmianę ze skanu I na skan II (t(14)?=??5.674, p<0.001, skan III (t(12) )?=??7.040, p<0.001 i skan IV (t(10)?=??3.258, p?=?0.009). Stwierdziliśmy również istotny wzrost podskali „ruchliwość fizyczna” od skanu I do skanu III (t(12)?=??3.974, p?=?0.002) i skan IV (t(10)?=??2.511, p?=?0.031).Nie było istotnej zmiany między tomem I a tomem II ( sześć tygodni po zabiegu).
Dane strukturalne
Analiza przekrojowa. Uwzględniliśmy wiek jako współzmienną w ogólnym modelu liniowym i nie znaleźliśmy żadnych sprzeczności związanych z wiekiem. W porównaniu z grupą kontrolną dopasowaną pod względem płci i wieku, pacjenci z pierwotną chorobą zwyrodnieniową stawu biodrowego (n?=~20) wykazywali przedoperacyjnie (Skana I) zmniejszoną ilość istoty szarej w przednim zakręcie obręczy (ACC), korze wyspy, wieczku, grzbietowo-bocznej korze przedczołowej ( DLPFC), prawy biegun skroniowy i móżdżek (tab. 1 i ryc. 1). Z wyjątkiem prawej skorupy (x?=?31, y?=??14, z?=??1; p<0.001, t?=?3.32) nie stwierdzono istotnego wzrostu gęstości istoty szarej u pacjentów z OA w porównaniu do zdrowych kontroli. Porównując pacjentów w punkcie czasowym IV skanu z dopasowanymi kontrolami, uzyskano te same wyniki, co w analizie przekrojowej z użyciem skanu I w porównaniu z grupą kontrolną.
Rycina 1: Statystyczne mapy parametryczne przedstawiające różnice strukturalne istoty szarej u pacjentów z przewlekłym bólem spowodowanym pierwotną chorobą zwyrodnieniową stawu biodrowego w porównaniu z grupą kontrolną i podłużnie w porównaniu z nimi w czasie. Znaczące zmiany istoty szarej pokazano nałożone na siebie kolorem, dane przekrojowe przedstawiono na czerwono, a dane podłużne na żółto. Płaszczyzna osiowa: lewa strona obrazu to lewa strona mózgu. u góry: Obszary znacznego zmniejszenia istoty szarej między pacjentami z przewlekłym bólem z powodu pierwotnej choroby zwyrodnieniowej stawu biodrowego i zdrowymi osobami z grupy kontrolnej. p<0.001 nieskorygowane dno: Wzrost istoty szarej u 20 pacjentów bez bólu w trzecim i czwartym okresie skanowania po całkowitej alloplastyce stawu biodrowego, w porównaniu z pierwszym (przedoperacyjnym) i drugim (6 tygodnia po zabiegu) skanem. p<8 nieskorygowane wykresy: szacunki kontrastu i 0.001% przedział ufności, efekty zainteresowania, jednostki arbitralne. oś x: kontrasty dla 90 punktów czasowych, oś y: ocena kontrastu przy a4, 3, 50 dla ACC i ocena kontrastu przy 2, 36, 39 dla wyspy.
Odwrócenie danych pacjentów z chorobą zwyrodnieniową lewego biodra (n?=?7) i porównanie ich ze zdrowymi kontrolami nie zmieniło znacząco wyników, ale dla zmniejszenia wzgórza (x?=?10, y?=??20, z?=?3, p<0.001, t?=?3.44) oraz wzrost prawego móżdżku (x?=?25, y?=??37, z?=??50, p<0.001, t? =<5.12), które nie osiągnęły istotności w nieodwróconych danych pacjentów w porównaniu z kontrolami.
Analiza podłużna. W analizie podłużnej wykryto istotny wzrost (nieskorygowany p<001) istoty szarej, porównując pierwszy i drugi skan (ból przewlekły/ból pooperacyjny) z trzecim i czwartym skanem (bez bólu) w ACC, kora wyspy, móżdżek i pars orbitalis u pacjentów z chorobą zwyrodnieniową stawów (tab. 2 i ryc. 1). Istota szara zmniejszyła się z czasem (p<001 analiza całego mózgu bez korekty) we wtórnej korze czuciowo-somatycznej, hipokampie, korze środkowej obręczy, wzgórzu i jądrze ogoniastym u pacjentów z chorobą zwyrodnieniową stawów (ryc. 2).
Rysunek 2: a) Znaczący wzrost istoty szarej mózgu po udanej operacji. Osiowy widok znacznego zmniejszenia istoty szarej u pacjentów z przewlekłym bólem z powodu pierwotnej choroby zwyrodnieniowej stawu biodrowego w porównaniu z osobnikami kontrolnymi. p<0.001 nieskorygowane (analiza przekrojowa), b) Wzdłużny wzrost istoty szarej w czasie w kolorze żółtym porównując skan I i IIscan III > skan IV) u pacjentów z chorobą zwyrodnieniową stawów. p<0.001 nieskorygowane (analiza podłużna). Lewa strona obrazu to lewa strona mózgu.
Odwrócenie danych pacjentów z OA lewego biodra (n?=?7) nie zmieniło znacząco wyników, ale dla zmniejszenia istoty szarej mózgu w Zakręcie Heschla (x?=??41, y?=?? 21, z?=?10, p<0.001, t?=?3.69) i przedklinek (x?=?15, y?=??36, z?=?3, p<0.001, t?=?4.60) .
Kontrastując pierwszy skan (przedoperacyjny) ze skanami 3+4 (pooperacyjnymi), stwierdziliśmy wzrost istoty szarej w korze czołowej i korze ruchowej (nieskorygowane p<0.001). Zauważamy, że ten kontrast jest mniej rygorystyczny, ponieważ mamy teraz mniej skanów na stan (ból w porównaniu z brakiem bólu). Kiedy obniżamy próg, powtarzamy to, co znaleźliśmy, używając kontrastu 1+2 vs. 3+4.
Szukając obszarów, które zwiększają się we wszystkich przedziałach czasowych, znaleźliśmy zmiany istoty szarej mózgu w obszarach motorycznych (obszar 6) u pacjentów ze zwyrodnieniem stawów biodrowych po całkowitej alloplastyce stawu biodrowego (skan Idbm.neuro.uni-jena.de/vbm/) moglibyśmy powtórzyć to odkrycie w przedniej i środkowej części zakrętu obręczy oraz na obu przednich wyspach.
Obliczyliśmy rozmiary efektu, a analiza przekrojowa (pacjenci vs kontrolni) dała Cohensd 1.78751 w piku wokselu ACC (x?=??12, y?=?25, z?=?? 16). Obliczyliśmy również współczynnik Cohena dla analizy podłużnej (przebieg kontrastowy 1+2 vs. skan 3+4). Spowodowało to, że współczynnik Cohensd wyniósł 1.1158 w ACC (x?=??3, y?=?50, z?=?2). W odniesieniu do wyspy (x?=??33, y?=?21, z?=?13) i w odniesieniu do tego samego kontrastu, Cohen�sd wynosi 1.0949. Dodatkowo obliczyliśmy średnią niezerowych wartości wokseli na mapie Cohensd w obrębie obszaru ROI (składającego się z przedniego podziału zakrętu obręczy i kory podproszkowej, pochodzących z Harvard-Oxford Cortical Structural Atlas): 1.251223.
Wgląd doktora Alexa Jimeneza
Pacjenci z przewlekłym bólem mogą z czasem doświadczać różnych problemów zdrowotnych, oprócz już wyniszczających objawów. Na przykład wiele osób doświadcza problemów ze snem w wyniku bólu, ale co najważniejsze, przewlekły ból może również prowadzić do różnych problemów ze zdrowiem psychicznym, w tym lęku i depresji. Skutki, jakie ból może mieć na mózg, mogą wydawać się zbyt przytłaczające, ale coraz więcej dowodów sugeruje, że te zmiany w mózgu nie są trwałe i można je odwrócić, gdy pacjenci z przewlekłym bólem otrzymają odpowiednie leczenie ich podstawowych problemów zdrowotnych. Według artykułu nieprawidłowości istoty szarej występujące w przewlekłym bólu nie odzwierciedlają uszkodzenia mózgu, ale są raczej odwracalną konsekwencją, która normalizuje się, gdy ból jest odpowiednio leczony. Na szczęście dostępnych jest wiele metod leczenia, które pomagają złagodzić objawy przewlekłego bólu oraz przywrócić strukturę i funkcję mózgu.
Dyskusja
Monitorując całą strukturę mózgu w czasie, potwierdzamy i rozszerzamy nasze niedawno opublikowane dane pilotażowe [17]. Znaleźliśmy zmiany w istocie szarej mózgu u pacjentów z pierwotną chorobą zwyrodnieniową stawu biodrowego w stanie przewlekłego bólu, które częściowo ustępują, gdy pacjenci ci nie odczuwają bólu po operacji endoprotezy stawu biodrowego. Częściowy wzrost istoty szarej po operacji występuje prawie w tych samych obszarach, w których przed operacją zaobserwowano spadek istoty szarej. Odwrócenie danych pacjentów z chorobą zwyrodnieniową stawu biodrowego lewego biodra (a tym samym normalizacja dla strony bólu) miało tylko niewielki wpływ na wyniki, ale dodatkowo wykazało zmniejszenie istoty szarej w zakręcie Heschla i Precuneus, którego nie możemy łatwo wytłumaczyć i, ponieważ nie istnieje żadna hipoteza a priori, należy zachować ostrożność. Jednak różnica obserwowana między pacjentami a osobami zdrowymi w skanie I była nadal widoczna w analizie przekrojowej w skanie IV. Względny wzrost istoty szarej w czasie jest zatem subtelny, tj. niewystarczająco wyraźny, aby mieć wpływ na analizę przekrojową, co zostało już wykazane w badaniach dotyczących plastyczności zależnej od doświadczenia [30], [31]. Zauważamy, że fakt, iż pokazujemy, że niektóre części zmian w mózgu spowodowane przewlekłym bólem są odwracalne, nie wyklucza, że niektóre inne części tych zmian są nieodwracalne.
Co ciekawe, zaobserwowaliśmy, że spadek istoty szarej w ACC u pacjentów z przewlekłym bólem przed operacją wydaje się utrzymywać 6 tygodni po operacji (skan II) i zwiększa się tylko do skanu III i IV, prawdopodobnie z powodu bólu pooperacyjnego lub zmniejszenia motoryki funkcjonować. Jest to zgodne z danymi behawioralnymi oceny mobilności fizycznej zawartej w NHP, która po operacji nie wykazała żadnej znaczącej zmiany w punkcie czasowym II, ale znacznie wzrosła w kierunku skanu III i IV. Warto zauważyć, że nasi pacjenci nie zgłaszali bólu w biodrze po operacji, ale odczuwali ból pooperacyjny w otaczających mięśniach i skórze, który był przez pacjentów bardzo różnie odbierany. Jednakże, ponieważ pacjenci nadal zgłaszali pewien ból w skanie II, porównaliśmy również pierwszy skan (przed operacją) ze skanami III+IV (po zabiegu), ujawniając wzrost istoty szarej w korze czołowej i korze ruchowej. Zauważamy, że ten kontrast jest mniej rygorystyczny z powodu mniejszej liczby skanów na stan (ból w porównaniu z brakiem bólu). Kiedy obniżyliśmy próg, powtarzamy to, co znaleźliśmy, używając kontrastu I+II vs. III+IV.
Nasze dane silnie sugerują, że zmiany istoty szarej u pacjentów z przewlekłym bólem, które zwykle występują w obszarach zaangażowanych w nadrdzeniowe przetwarzanie nocyceptywne [4], nie są spowodowane atrofią neuronów ani uszkodzeniem mózgu. Fakt, że te zmiany obserwowane w stanie bólu przewlekłego nie ustępują całkowicie, można wytłumaczyć stosunkowo krótkim okresem obserwacji (rok po operacji w porównaniu ze średnią siedmiu lat bólu przewlekłego przed operacją). Zmiany neuroplastyczne w mózgu, które mogły rozwinąć się przez kilka lat (w konsekwencji ciągłego bodźca nocyceptywnego) wymagają prawdopodobnie więcej czasu, aby całkowicie się cofnąć. Inną możliwością, dlaczego wzrost istoty szarej można wykryć tylko w danych podłużnych, ale nie w danych przekrojowych (tj. między kohortami w punkcie czasowym IV), jest to, że liczba pacjentów (n?=?20) jest zbyt mała. Należy podkreślić, że wariancja między mózgami kilku osób jest dość duża, a dane podłużne mają tę zaletę, że wariancja jest stosunkowo niewielka, ponieważ te same mózgi są skanowane kilkakrotnie. W konsekwencji subtelne zmiany będą wykrywalne tylko w danych podłużnych [30], [31], [32]. Oczywiście nie możemy wykluczyć, że zmiany te są przynajmniej częściowo nieodwracalne, chociaż jest to mało prawdopodobne, biorąc pod uwagę ustalenia dotyczące plastyczności strukturalnej i reorganizacji specyficznej dla ćwiczeń [4], [12], [30], [33], [34]. Aby odpowiedzieć na to pytanie, przyszłe badania muszą wielokrotnie badać pacjentów w dłuższych ramach czasowych, być może lat.
Zauważamy, że możemy wyciągnąć jedynie ograniczone wnioski dotyczące dynamiki zmian morfologicznych mózgu w czasie. Powodem jest to, że kiedy projektowaliśmy to badanie w 2007 r. i skanowaliśmy w 2008 i 2009 r., nie było wiadomo, czy zmiany strukturalne w ogóle wystąpią, i ze względu na wykonalność wybraliśmy daty skanowania i ramy czasowe, jak opisano tutaj. Można argumentować, że zmiany istoty szarej w czasie, które opisujemy dla grupy pacjentów, mogły mieć miejsce również w grupie kontrolnej (efekt czasu). Oczekuje się jednak, że wszelkie zmiany spowodowane starzeniem się, jeśli w ogóle, będą spadkiem wolumenu. Biorąc pod uwagę naszą hipotezę a priori, opartą na 9 niezależnych badaniach i kohortach wykazujących zmniejszenie istoty szarej u pacjentów z przewlekłym bólem [7], [8], [9], [15], [24], [25], [26], [27], [28], skupiliśmy się na regionalnych wzrostach w czasie i dlatego uważamy, że nasze odkrycie nie jest prostym efektem czasowym. Należy zauważyć, że nie możemy wykluczyć, że spadek istoty szarej w czasie, który stwierdziliśmy w naszej grupie pacjentów, może być spowodowany efektem czasu, ponieważ nie skanowaliśmy naszej grupy kontrolnej w tym samym przedziale czasowym. Biorąc pod uwagę wyniki, przyszłe badania powinny mieć na celu większą liczbę krótszych odstępów czasowych, biorąc pod uwagę, że zależne od wysiłku zmiany morfometryczne mózgu mogą wystąpić już po 1 tygodniu [32], [33].
Oprócz wpływu nocyceptywnego aspektu bólu na istotę szarą mózgu [17], [34] zaobserwowaliśmy, że zmiany funkcji motorycznych prawdopodobnie również przyczyniają się do zmian strukturalnych. Odkryliśmy, że obszary motoryczne i przedruchowe (obszar 6) zwiększają się we wszystkich przedziałach czasowych (ryc. 3). Intuicyjnie może to być spowodowane poprawą funkcji motorycznych w czasie, ponieważ pacjenci nie byli już bardziej ograniczani w prowadzeniu normalnego życia. W szczególności nie skupiliśmy się na funkcji motorycznej, ale na poprawie odczuwania bólu, biorąc pod uwagę nasze pierwotne dążenie do zbadania, czy dobrze znana redukcja istoty szarej mózgu u pacjentów z przewlekłym bólem jest w zasadzie odwracalna. W związku z tym nie używaliśmy specjalnych narzędzi do badania funkcji motorycznych. Niemniej jednak (funkcjonalna) reorganizacja kory ruchowej u pacjentów z zespołami bólowymi jest dobrze udokumentowana [35], [36], [37], [38]. Co więcej, kora ruchowa jest jednym z celów terapeutycznych u pacjentów z przewlekłym bólem nieuleczalnym medycznie, stosujących bezpośrednią stymulację mózgu [39], [40], przezczaszkową stymulację prądem stałym [41] i powtarzalną przezczaszkową stymulację magnetyczną [42], [43]. Dokładne mechanizmy takiej modulacji (ułatwianie vs hamowanie lub po prostu ingerencja w sieci związane z bólem) nie są jeszcze wyjaśnione [40]. Niedawne badania wykazały, że określone doświadczenie motoryczne może zmienić strukturę mózgu [13]. Synaptogeneza, reorganizacja reprezentacji ruchu i angiogeneza w korze ruchowej mogą wystąpić przy specjalnych wymaganiach zadania ruchowego. Tsao i in. wykazali reorganizację w korze ruchowej pacjentów z przewlekłym bólem krzyża, który wydaje się być specyficzny dla bólu pleców [44], a Puri i wsp. zaobserwowali zmniejszenie lewego uzupełniającego obszaru ruchowego istoty szarej u osób cierpiących na fibromialgię [45]. Nasze badanie nie zostało zaprojektowane w celu rozwikłania różnych czynników, które mogą zmieniać mózg w przewlekłym bólu, ale interpretujemy nasze dane dotyczące zmian istoty szarej, że nie odzwierciedlają one wyłącznie konsekwencji ciągłego bodźca nocyceptywnego. W rzeczywistości niedawne badanie pacjentów z bólem neuropatycznym wykazało nieprawidłowości w obszarach mózgu obejmujących emocjonalną, autonomiczną i percepcję bólu, co sugeruje, że odgrywają one kluczową rolę w globalnym obrazie klinicznym przewlekłego bólu [28].
Rycina 3: Statystyczne mapy parametryczne wykazujące znaczny wzrost istoty szarej mózgu w obszarach motorycznych (obszar 6) u pacjentów z chorobą zwyrodnieniową stawów przed w porównaniu z po THR (analiza podłużna, skan I Oszacowania kontrastu przy x?=?19, y?=??12, z?=?70.
Dwa niedawne badania pilotażowe skupiały się na terapii endoprotezoplastyki stawu biodrowego u pacjentów z chorobą zwyrodnieniową stawów, jedynym przewlekłym zespołem bólowym, który można głównie wyleczyć za pomocą całkowitej alloplastyki stawu biodrowego [17], [46], a dane te są poprzedzone bardzo niedawnym badaniem u pacjentów z przewlekłym bólem krzyża [ 47]. Badania te należy rozpatrywać w świetle kilku badań podłużnych badających zależną od doświadczenia plastyczność neuronalną u ludzi na poziomie strukturalnym [30], [31] oraz niedawne badania dotyczące strukturalnych zmian mózgu u zdrowych ochotników doświadczających powtarzającej się bolesnej stymulacji [34]. . Kluczowym przesłaniem wszystkich tych badań jest to, że główna różnica w strukturze mózgu między pacjentami cierpiącymi na ból a grupą kontrolną może zanikać, gdy ból zostanie wyleczony. Należy jednak wziąć pod uwagę, że po prostu nie jest jasne, czy zmiany u pacjentów z przewlekłym bólem wynikają wyłącznie z bodźców nocyceptywnych, czy z konsekwencji bólu, czy też obu. Jest więcej niż prawdopodobne, że zmiany behawioralne, takie jak deprywacja lub wzmocnienie kontaktów społecznych, zwinność, trening fizyczny i zmiany stylu życia wystarczą do ukształtowania mózgu [6], [12], [28], [48]. Szczególnie depresja jako choroba współistniejąca lub konsekwencja bólu jest kluczowym kandydatem do wyjaśnienia różnic między pacjentami a grupą kontrolną. Niewielka grupa naszych pacjentów z chorobą zwyrodnieniową stawów wykazała łagodne do umiarkowanych objawy depresji, które zmieniały się w czasie. Nie stwierdziliśmy, że zmiany strukturalne istotnie współzmienne z wynikiem BDI, ale powstaje pytanie, ile innych zmian behawioralnych spowodowanych brakiem bólu i poprawą motoryczną może przyczynić się do wyników iw jakim stopniu. Te zmiany behawioralne mogą prawdopodobnie wpływać na zmniejszenie istoty szarej w przewlekłym bólu, a także na wzrost istoty szarej, gdy ból zniknie.
Innym ważnym czynnikiem, który może zafałszować naszą interpretację wyników, jest fakt, że prawie wszyscy pacjenci z przewlekłym bólem przyjmowali leki przeciwbólowe, które odstawiali, gdy nie odczuwali bólu. Można argumentować, że NLPZ, takie jak diklofenak lub ibuprofen, mają pewien wpływ na układ nerwowy i to samo dotyczy opioidów, leków przeciwpadaczkowych i antydepresyjnych, leków często stosowanych w leczeniu przewlekłego bólu. Wpływ środków przeciwbólowych i innych leków na wyniki morfometryczne może być równie ważny (48). Żadne z dotychczasowych badań nie wykazało wpływu leków przeciwbólowych na morfologię mózgu, ale w kilku pracach stwierdzono, że zmiany w strukturze mózgu u pacjentów z przewlekłym bólem nie można wyjaśnić wyłącznie brakiem aktywności związanej z bólem [15], ani lekiem przeciwbólowym [7], [9]. [49]. Brakuje jednak konkretnych badań. Dalsze badania powinny koncentrować się na zależnych od doświadczenia zmianach plastyczności kory mózgowej, które mogą mieć ogromne implikacje kliniczne w leczeniu przewlekłego bólu.
W analizie podłużnej stwierdziliśmy również zmniejszenie istoty szarej, prawdopodobnie z powodu procesów reorganizacji, które towarzyszą zmianom funkcji motorycznych i percepcji bólu. Niewiele jest dostępnych informacji na temat zmian podłużnych istoty szarej mózgu w stanach bólowych, dlatego nie mamy hipotezy o spadku istoty szarej w tych obszarach po operacji. Teutsch i in. [25] stwierdzili wzrost istoty szarej mózgu w korze somatosensorycznej i środkowej części zakrętu obręczy u zdrowych ochotników, którzy doświadczali bolesnej stymulacji w codziennym protokole przez osiem kolejnych dni. Stwierdzenie wzrostu istoty szarej po eksperymentalnym bodźcu nocyceptywnym pokrywało się w pewnym stopniu anatomicznie ze spadkiem istoty szarej mózgu w tym badaniu u pacjentów, którzy zostali wyleczeni z długotrwałego przewlekłego bólu. Oznacza to, że bodźce nocyceptywne u zdrowych ochotników prowadzą do zmian strukturalnych zależnych od wysiłku, jak to prawdopodobnie ma miejsce u pacjentów z przewlekłym bólem, i że zmiany te odwracają się u zdrowych ochotników, gdy ustają bodźce nocyceptywne. W konsekwencji, zmniejszenie istoty szarej w tych obszarach obserwowane u pacjentów z ChZS można interpretować jako podążanie za tym samym podstawowym procesem: zmiany zależne od wysiłku, zmiany w mózgu [50]. Jako procedura nieinwazyjna, Morfometria MR jest idealnym narzędziem do poszukiwania morfologicznych substratów chorób, pogłębiania naszego zrozumienia związku między strukturą a funkcją mózgu, a nawet monitorowania interwencji terapeutycznych. Jednym z największych wyzwań w przyszłości jest przystosowanie tego potężnego narzędzia do wieloośrodkowych i terapeutycznych badań bólu przewlekłego.
Ograniczenia tego badania
Chociaż to badanie jest rozszerzeniem naszego poprzedniego badania, rozszerzając dane z obserwacji do 12 miesięcy i badając większą liczbę pacjentów, nasze główne odkrycie, że zmiany morfometryczne mózgu w przewlekłym bólu są odwracalne, jest raczej subtelne. Wielkości efektów są małe (patrz powyżej), a efekty są częściowo napędzane przez dalsze zmniejszenie objętości regionalnej istoty szarej mózgu w punkcie czasowym skanu 2. Gdy wykluczymy dane ze skanu 2 (bezpośrednio po operacji) tylko znaczące wzrost istoty szarej mózgu w korze ruchowej i korze czołowej przeżywa p<0.001 nieskorygowany (Tabela 3).
Wnioski
Nie jest możliwe rozróżnienie, w jakim stopniu obserwowane przez nas zmiany strukturalne są spowodowane zmianami w bodźcach nocyceptywnych, zmianami funkcji motorycznych lub przyjmowaniem leków lub zmianami w samopoczuciu. Zamaskowanie kontrastów grupowych pierwszego i ostatniego skanu między sobą ujawniło znacznie mniej różnic niż oczekiwano. Przypuszczalnie zmiany w mózgu spowodowane przewlekłym bólem ze wszystkimi konsekwencjami rozwijają się przez dość długi czas i mogą również potrzebować trochę czasu, aby powrócić. Niemniej jednak wyniki te ujawniają procesy reorganizacji, silnie sugerując, że przewlekłe bodźce nocyceptywne i upośledzenie motoryczne u tych pacjentów prowadzi do zmienionego przetwarzania w obszarach korowych, aw konsekwencji strukturalnych zmian mózgu, które są w zasadzie odwracalne.
Podziękowanie
Dziękujemy wszystkim wolontariuszom za udział w tym badaniu oraz grupie Fizyki i Metod w NeuroImage Nord w Hamburgu. Badanie zostało zatwierdzone przez lokalną komisję ds. etyki, a przed badaniem uzyskano pisemną świadomą zgodę od wszystkich uczestników badania.
Oświadczenie o finansowaniu
Praca ta była finansowana z grantów DFG (Niemieckiej Fundacji Badawczej) (MA 1862/2-3) i BMBF (Federalne Ministerstwo Edukacji i Badań) (371 57 01 i NeuroImage Nord). Fundatorzy nie brali udziału w projektowaniu badań, gromadzeniu i analizowaniu danych, podejmowaniu decyzji o publikacji czy przygotowaniu rękopisu.
Układ endokannabinoidowy: niezbędny układ, o którym nigdy nie słyszałeś
Jeśli nie słyszałeś o systemie endokannabinoidowym lub ECS, nie musisz się wstydzić. W latach sześćdziesiątych badacze, którzy zainteresowali się bioaktywnością konopi, w końcu wyizolowali wiele z jej aktywnych substancji chemicznych. Jednak badaczom badającym modele zwierzęce zajęło kolejne 1960 lat znalezienie receptora dla tych substancji chemicznych ECS w mózgach gryzoni, odkrycie, które otworzyło cały świat badań nad istnieniem receptorów ECS i ich fizjologicznym przeznaczeniem.
Teraz wiemy, że większość zwierząt, od ryb przez ptaki po ssaki, posiada endokannabinoidy i wiemy, że ludzie nie tylko wytwarzają własne kannabinoidy, które wchodzą w interakcje z tym konkretnym układem, ale także wytwarzamy inne związki, które oddziałują z ECS, te z które są obserwowane w wielu różnych roślinach i żywności, daleko poza gatunkami konopi.
Jako system ludzkiego ciała ECS nie jest wyizolowaną platformą strukturalną, taką jak układ nerwowy czy układ sercowo-naczyniowy. Zamiast tego ECS to zestaw receptorów szeroko rozpowszechnionych w całym ciele, które są aktywowane przez zestaw ligandów, które wspólnie znamy jako endokannabinoidy lub kannabinoidy endogenne. Oba zweryfikowane receptory są nazywane po prostu CB1 i CB2, chociaż istnieją inne, które zostały zaproponowane. Kanały PPAR i TRP również pośredniczą w niektórych funkcjach. Podobnie znajdziesz tylko dwa dobrze udokumentowane endokannabinoidy: anadamid i 2-arachidonoiloglicerol, czyli 2-AG.
Co więcej, fundamentalne dla układu endokannabinoidowego są enzymy, które syntetyzują i rozkładają endokannabinoidy. Uważa się, że endokannabinoidy są syntetyzowane w razie potrzeby. Głównymi zaangażowanymi enzymami są lipaza diacyloglicerolowa i N-acylo-fosfatydyloetanoloamino-fosfolipaza D, które odpowiednio syntetyzują 2-AG i anandamid. Dwa główne enzymy degradujące to hydrolaza amidów kwasów tłuszczowych (FAAH), która rozkłada anandamid, oraz lipaza monoacyloglicerolowa (MAGL), która rozkłada 2-AG. Regulacja tych dwóch enzymów może zwiększać lub zmniejszać modulację ECS.
Jaka jest funkcja ECS?
ECS jest głównym homeostatycznym systemem regulacyjnym organizmu. Można go łatwo postrzegać jako wewnętrzny system adaptogenny organizmu, zawsze działający na rzecz utrzymania równowagi różnych funkcji. Endokannabinoidy działają szeroko jako neuromodulatory i jako takie regulują szeroki zakres procesów zachodzących w organizmie, od płodności po ból. Oto niektóre z tych lepiej znanych funkcji ECS:
System nerwowy
Z ośrodkowego układu nerwowego lub OUN ogólna stymulacja receptorów CB1 będzie hamować uwalnianie glutaminianu i GABA. W OUN ECS odgrywa rolę w tworzeniu i uczeniu się pamięci, promuje neurogenezę w hipokampie, reguluje również pobudliwość neuronalną. ECS odgrywa również rolę w sposobie, w jaki mózg reaguje na urazy i stany zapalne. Z rdzenia kręgowego ECS moduluje sygnalizację bólu i wzmacnia naturalną analgezję. W obwodowym układzie nerwowym, w którym kontrolują receptory CB2, ECS działa przede wszystkim we współczulnym układzie nerwowym, regulując funkcje przewodu pokarmowego, moczowego i rozrodczego.
Stres i nastrój
ECS ma wieloraki wpływ na reakcje na stres i regulację emocjonalną, taki jak inicjacja reakcji organizmu na ostry stres i adaptacja w czasie do bardziej długotrwałych emocji, takich jak strach i niepokój. Zdrowo działający system endokannabinoidowy ma kluczowe znaczenie dla tego, jak ludzie modulują między satysfakcjonującym stopniem pobudzenia a poziomem, który jest nadmierny i nieprzyjemny. ECS odgrywa również rolę w tworzeniu pamięci, a być może zwłaszcza w sposobie, w jaki mózg zapisuje wspomnienia ze stresu lub urazu. Ponieważ ECS moduluje uwalnianie dopaminy, noradrenaliny, serotoniny i kortyzolu, może również wpływać na reakcje emocjonalne i zachowania.
Układ trawienny
Przewód pokarmowy jest wypełniony zarówno receptorami CB1, jak i CB2, które regulują kilka ważnych aspektów zdrowia przewodu pokarmowego. Uważa się, że ECS może być „brakującym ogniwem” w opisie połączenia jelitowo-mózgowo-odpornościowego, które odgrywa znaczącą rolę w funkcjonalnym zdrowiu przewodu pokarmowego. ECS jest regulatorem odporności jelitowej, być może poprzez ograniczenie niszczenia zdrowej flory przez układ odpornościowy, a także poprzez modulację sygnalizacji cytokin. ECS moduluje naturalną odpowiedź zapalną w przewodzie pokarmowym, co ma ważne implikacje dla wielu problemów zdrowotnych. Wydaje się, że motoryka żołądka i ogólnego układu pokarmowego również jest częściowo regulowana przez ECS.
Apetyt i metabolizm
ECS, w szczególności receptory CB1, odgrywają rolę w apetycie, metabolizmie i regulacji tkanki tłuszczowej. Stymulacja receptorów CB1 wzmaga zachowania związane z poszukiwaniem pokarmu, poprawia świadomość węchu, reguluje również bilans energetyczny. Zarówno zwierzęta, jak i ludzie z nadwagą mają zaburzenia ECS, które mogą prowadzić do nadaktywności tego układu, co przyczynia się zarówno do przejadania się, jak i zmniejszenia wydatku energetycznego. Wykazano, że poziomy anandamidu i 2-AG w krążeniu są podwyższone w przypadku otyłości, co może być częściowo spowodowane zmniejszoną produkcją enzymu rozkładającego FAAH.
Zdrowie immunologiczne i reakcja zapalna
Komórki i narządy układu odpornościowego są bogate w receptory endokannabinoidowe. Receptory kannabinoidowe są wyrażane w grasicy, śledzionie, migdałkach i szpiku kostnym, a także na limfocytach T i B, makrofagach, komórkach tucznych, neutrofilach i komórkach NK. ECS jest uważany za główny czynnik odpowiedzialny za równowagę i homeostazę układu odpornościowego. Chociaż nie wszystkie funkcje ECS z układu odpornościowego są zrozumiałe, wydaje się, że ECS reguluje produkcję cytokin, a także odgrywa rolę w zapobieganiu nadaktywności układu odpornościowego. Zapalenie jest naturalną częścią odpowiedzi immunologicznej i odgrywa bardzo normalną rolę w ostrych urazach ciała, w tym urazach i chorobach; niemniej jednak, gdy nie jest utrzymywany w ryzach, może stać się przewlekły i przyczynić się do kaskady niekorzystnych problemów zdrowotnych, takich jak przewlekły ból. Utrzymując odpowiedź immunologiczną w ryzach, ECS pomaga utrzymać bardziej zrównoważoną odpowiedź zapalną w organizmie.
Inne obszary zdrowia regulowane przez ECS:
Zdrowia kości
Płodność
zdrowie skóry
Zdrowie tętnic i dróg oddechowych
Sen i rytm dobowy
Jak najlepiej wspierać zdrowy ECS to pytanie, na które obecnie stara się odpowiedzieć wielu badaczy. Bądź na bieżąco, aby uzyskać więcej informacji na ten pojawiający się temat.
PodsumowującPrzewlekły ból jest związany ze zmianami w mózgu, w tym redukcją istoty szarej. Jednak powyższy artykuł wykazał, że przewlekły ból może zmienić ogólną strukturę i funkcję mózgu. Chociaż przewlekły ból może prowadzić do tych, między innymi problemów zdrowotnych, właściwe leczenie podstawowych objawów pacjenta może odwrócić zmiany w mózgu i regulować istotę szarą. Co więcej, coraz więcej badań naukowych ujawniło znaczenie systemu endokannabinoidowego i jego funkcji w kontrolowaniu, a także radzeniu sobie z przewlekłym bólem i innymi problemami zdrowotnymi. Informacje podane w National Center for Biotechnology Information (NCBI). Zakres naszych informacji ogranicza się do chiropraktyki oraz urazów i schorzeń kręgosłupa. Aby omówić ten temat, prosimy zapytać dr Jimeneza lub skontaktować się z nami pod adresem�915-850-0900 .
Kurator: dr Alex Jimenez
Dodatkowe tematy: Back Pain
Na ból pleców jest jedną z najczęstszych przyczyn niepełnosprawności i nieobecności w pracy na całym świecie. W rzeczywistości, ból pleców został przypisany jako drugi najczęstszy powód wizyt u lekarza, przewyższający jedynie infekcje górnych dróg oddechowych. Około 80 procent populacji doświadczy pewnego rodzaju bólu pleców przynajmniej raz w ciągu całego życia. Kręgosłup jest złożoną strukturą zbudowaną z kości, stawów, więzadeł i mięśni oraz innych miękkich tkanek. Z tego powodu urazy i / lub pogorszenie warunków, takich jak przepukliny, może ostatecznie prowadzić do objawów bólu pleców. Urazy sportowe lub wypadki samochodowe są często najczęstszą przyczyną bólu pleców, jednak czasami najprostsze ruchy mogą mieć bolesne skutki. Na szczęście alternatywne opcje leczenia, takie jak opieka chiropraktyczna, mogą pomóc złagodzić ból pleców poprzez zastosowanie korekcji kręgosłupa i manualnych manipulacji, ostatecznie poprawiając ulgę w bólu.
3.�Wrigley PJ, Gustin SM, Macey PM, Nash PG, Gandevia SC i in. (2009)�Zmiany anatomiczne w korze ruchowej człowieka i drogach ruchowych po całkowitym uszkodzeniu rdzenia kręgowego w odcinku piersiowym. Cereb Cortex19: 224.�[PubMed]
4.�Maj A (2008)�Przewlekły ból może zmienić strukturę mózgu. Ból137: 7.�[PubMed]
5.�Maj A (2009) Morfowanie wokseli: szum wokół obrazowania strukturalnego pacjentów z bólami głowy. Mózg.[PubMed]
7.�Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN i in. (2004)�Przewlekły ból pleców jest związany ze zmniejszoną gęstością istoty szarej przedczołowej i wzgórzowej. J Neurosci24: 10410.�[PubMed]
8.�Rocca MA, Ceccarelli A, Falini A, Colombo B, Tortorella P i in. (2006)�Zmiany istoty szarej mózgu u pacjentów z migreną z widocznymi zmianami w obrazach T2: badanie 3-T MRI. Uderzenie37: 1765.�[PubMed]
9.�Kuchinad A, Schweinhardt P, Seminowicz DA, Wood PB, Chizh BA i in. (2007)�Przyspieszona utrata istoty szarej mózgu u pacjentów z fibromialgią: przedwczesne starzenie się mózgu?J Neurosci27: 4004.[PubMed]
10.�Tracey I, Bushnell MC (2009).W jaki sposób badania neuroobrazowe skłoniły nas do ponownego przemyślenia: czy przewlekły ból jest chorobą?J Ból10: 1113.�[PubMed]
11.�Franke K, Ziegler G, Kloppel S, Gaser C (2010).Szacowanie wieku zdrowych osób na podstawie skanów T1-zależnych MRI przy użyciu metod jądra: badanie wpływu różnych parametrów. Neuroimage50: 883.�[PubMed]
12.�Draganski B, maj A (2008)�Zmiany strukturalne wywołane treningiem w mózgu dorosłego człowieka. Behav Brain Res192: 137.�[PubMed]
13.�Adkins DL, Boychuk J, Remple MS, Kleim JA (2006).Trening motoryczny wywołuje charakterystyczne dla doświadczenia wzorce plastyczności w korze ruchowej i rdzeniu kręgowym. J Appl Physiol101: 1776.�[PubMed]
14.�Duerden EG, Laverdure-Dupont D (2008).Praktyka tworzy korę. J Neurosci28: 8655.�[PubMed]
15.�Draganski B, Moser T, Lummel N, Ganssbauer S, Bogdahn U i in. (2006)�Zmniejszenie szarej istoty wzgórza po amputacji kończyny. Neuroimage31: 951.�[PubMed]
16.�Nikolajsen L, Brandsborg B, Lucht U, Jensen TS, Kehlet H (2006)�Przewlekły ból po całkowitej alloplastyce stawu biodrowego: ogólnokrajowe badanie ankietowe. Acta Anestezjol Scand50: 495.�[PubMed]
17.�Rodriguez-Raecke R, Niemeier A, Ihle K, Ruether W, maj A (2009).Spadek istoty szarej mózgu w przewlekłym bólu jest konsekwencją, a nie przyczyną bólu. J Neurosci29: 13746.�[PubMed]
18.�Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J (1961)�Inwentarz do pomiaru depresji. Arch Gen Psychiatry4: 561.�[PubMed]
19.�Franke G (2002) Die Symptom-Checkliste nach LR Derogatis – Manual. Getynga Beltz Test Verlag.
20.�Geissner E (1995) The Pain Perception Scale – zróżnicowana i wrażliwa na zmiany skala do oceny bólu przewlekłego i ostrego. Rehabilitacja (Stuttg) 34: XXXV�XLIII.�[PubMed]
21.�Bullinger M, Kirchberger I (1998) SF-36 – Fragebogen zum Gesundheitszustand. Ręcznie anweisung. Getynga: Hogrefe.
23.�Dobra płyta CD, Johnsrude IS, Ashburner J, Henson RN, Friston KJ i in. (2001)�Morfometryczne badanie starzenia się 465 zdrowych mózgów dorosłych ludzi na podstawie wokseli. Neuroimage14: 21.�[PubMed]
24.�Baliki MN, Chialvo DR, Geha PY, Levy RM, Harden RN i in. (2006)�Ból przewlekły a mózg emocjonalny: specyficzna aktywność mózgu związana ze spontanicznymi wahaniami intensywności przewlekłego bólu pleców. J Neurosci26: 12165.�[Artykuł bezpłatny PMC][PubMed]
25.�Lutz J, Jager L, de Quervain D, Krauseneck T, Padberg F i in. (2008)�Nieprawidłowości istoty białej i szarej w mózgu pacjentów z fibromialgią: badanie z napinaczem dyfuzji i obrazowaniem wolumetrycznym. Zapalenie stawów58: 3960.�[PubMed]
26.�Wrigley PJ, Gustin SM, Macey PM, Nash PG, Gandevia SC i in. (2008)�Zmiany anatomiczne w korze ruchowej człowieka i szlakach ruchowych po całkowitym urazie rdzenia kręgowego w odcinku piersiowym. Cereb Cortex19: 224.�[PubMed]
27.�Schmidt-Wilcke T, Hierlmeier S, Leinisch E (2010) Zmieniona regionalna morfologia mózgu u pacjentów z przewlekłym bólem twarzy. Ból głowy.�[PubMed]
28.�Geha PY, Baliki MN, Harden RN, Bauer WR, Parrish TB, i in. (2008)�Mózg w przewlekłym bólu CRPS: nieprawidłowe interakcje istoty szarej z białą w regionach emocjonalnych i autonomicznych. Neuron60: 570.�[Artykuł bezpłatny PMC][PubMed]
29.�Brazier J, Roberts J, Deverill M (2002).Oszacowanie miary zdrowia opartej na preferencjach na podstawie SF-36. J Zdrowie Ekon21: 271.�[PubMed]
30.�Draganski B, Gaser C, Busch V, Schuierer G, Bogdahn U i in. (2004)�Neuroplastyczność: zmiany w istocie szarej wywołane treningiem. Natura427: 311.�[PubMed]
31.�Boyke J, Driemeyer J, Gaser C, Buchel C, maj A (2008)�Zmiany w strukturze mózgu wywołane treningami u osób starszych. J Neurosci28: 7031.�[PubMed]
32.�Driemeyer J, Boyke J, Gaser C, Buchel C, maj A (2008)�Zmiany w istocie szarej wywołane przez uczenie się�ponownie. PLoS ONE3: e2669.�[Artykuł bezpłatny PMC][PubMed]
33.�May A, Hajak G, Ganssbauer S, Steffens T, Langguth B i in. (2007)�Zmiany strukturalne mózgu po 5 dniach interwencji: dynamiczne aspekty neuroplastyczności. Cereb Cortex17: 205.�[PubMed]
34.�Teutsch S, Herken W, Bingel U, Schoell E, maj A (2008)�Zmiany w istocie szarej mózgu spowodowane powtarzającą się bolesną stymulacją. Neuroimage42: 845.�[PubMed]
35.�Flor H, Braun C, Elbert T, Birbaumer N (1997).Rozległa reorganizacja pierwotnej kory somatosensorycznej u pacjentów z przewlekłym bólem pleców. Neurosci Lett224: 5.�[PubMed]
36.�Flor H, Denke C, Schaefer M, Grusser S (2001).Wpływ treningu dyskryminacji sensorycznej na reorganizację korową i ból fantomowy kończyn. Lancet357: 1763.�[PubMed]
37.�Swart CM, Stins JF, Beek PJ (2009).Zmiany korowe w złożonym regionalnym zespole bólowym (CRPS). Eur J Pain13: 902.�[PubMed]
38.�Maihofner C, Baron R, DeCol R, Binder A, Birklein F i in. (2007)�Układ ruchu wykazuje zmiany adaptacyjne w złożonym regionalnym zespole bólowym. Mózg130: 2671.�[PubMed]
39.�Fontaine D, Hamani C, Lozano A (2009).Skuteczność i bezpieczeństwo stymulacji kory ruchowej w przewlekłym bólu neuropatycznym: krytyczny przegląd literatury. J Neurosurg110: 251.�[PubMed]
40.�Levy R, Jeleń TR, Henderson J (2010)�Neurostymulacja wewnątrzczaszkowa w celu kontroli bólu: przegląd. Lekarz bólu13: 157.�[PubMed]
41.�Antal A, Brepohl N, Poreisz C, Boros K, Csifcsak G i in. (2008)�Przezczaszkowa stymulacja prądem stałym nad korą somatosensoryczną zmniejsza doświadczalnie wywołane ostre odczuwanie bólu. Clin J Ból24: 56.�[PubMed]
42.�Teepker M, Hotzel J, Timmesfeld N, Reis J, Mylius V i in. (2010)�rTMS o niskiej częstotliwości wierzchołka w profilaktycznym leczeniu migreny. Cephalalgia30: 137.�[PubMed]
43.�O�Connell N, Różdżka B, Marston L, Spencer S, Desouza L (2010)�Nieinwazyjne techniki stymulacji mózgu w przypadku bólu przewlekłego. Raport z przeglądu systematycznego i metaanalizy Cochrane. Eur J Phys Rehabilitation Med47: 309.�[PubMed]
44.�Tsao H, Galea MP, Hodges PW (2008).Reorganizacja kory ruchowej wiąże się z deficytami kontroli postawy w nawracających bólach krzyża. Mózg131: 2161.�[PubMed]
45.�Puri BK, Agour M, Gunatilake KD, Fernando KA, Gurusinghe AI i in. (2010)�Zmniejszenie lewego dodatkowego obszaru motorycznego istoty szarej u dorosłych kobiet cierpiących na fibromialgię z wyraźnym zmęczeniem i bez zaburzeń afektywnych: pilotażowe badanie morfometrii opartej na obrazowaniu rezonansem magnetycznym 3T z użyciem wokseli. J Int Med Res38: 1468.�[PubMed]
46.�Gwilym SE, Filipini N, Douaud G, Carr AJ, Tracey I (2010) Zanik wzgórza związany z bolesną chorobą zwyrodnieniową stawu biodrowego jest odwracalny po endoprotezoplastyce; podłużne badanie morfometryczne oparte na wokselach. Zapalenie stawów.�[PubMed]
47.�Seminowicz DA, Wideman TH, Naso L, Hatami-Khoroushahi Z, Fallatah S i in. (2011)�Skuteczne leczenie przewlekłego bólu krzyża u ludzi odwraca nieprawidłową anatomię i funkcję mózgu. J Neurosci31: 7540.�[PubMed]
48.�Maj A, Gaser C (2006)�Morfometria oparta na rezonansie magnetycznym: okno na plastyczność strukturalną mózgu. Curr Opin Neurol19: 407.�[PubMed]
49.�Schmidt-Wilcke T, Leinisch E, Straube A, Kampfe N, Draganski B i in. (2005)�Zmniejszenie ilości szarej substancji u pacjentów z przewlekłym napięciowym bólem głowy. Neurologia65: 1483.�[PubMed]
50.�Maj A (2009)�Morphing voxels: szum wokół obrazowania strukturalnego pacjentów z bólami głowy. Mózg 132 (Pt6): 1419.�[PubMed]
Biochemia bólu:„Wszystkie zespoły bólowe mają profil zapalny. Profil stanu zapalnego może się różnić w zależności od osoby, a także może różnić się u jednej osoby w różnym czasie. Leczenie zespołów bólowych polega na zrozumieniu tego profilu zapalnego. Zespoły bólowe są leczone medycznie, chirurgicznie lub jedno i drugie. Celem jest zahamowanie/zahamowanie produkcji mediatorów stanu zapalnego. A pomyślny wynik to taki, który skutkuje mniejszym stanem zapalnym i oczywiście mniejszym bólem.
Biochemia bólu
Cele:
Kim są główni gracze?
Jakie są mechanizmy biochemiczne?
Jakie są konsekwencje?
Przegląd stanu zapalnego:
Kluczowi gracze
Dlaczego boli mnie ramię? Przegląd neuroanatomicznej i biochemicznej podstawy bólu barku
ABSTRACT
Jeśli pacjent zapyta „dlaczego boli mnie ramię?”, rozmowa szybko przerodzi się w teorię naukową, a czasem bezpodstawne domysły. Często klinicysta uświadamia sobie ograniczenia naukowej podstawy ich wyjaśnienia, wykazując niekompletność naszego rozumienia natury bólu barku. W niniejszym przeglądzie przyjęto systematyczne podejście, aby pomóc odpowiedzieć na podstawowe pytania dotyczące bólu barku, z myślą o zapewnieniu wglądu w przyszłe badania i nowe metody leczenia bólu barku. Zbadamy role (1) receptorów obwodowych, (2) przetwarzania bólu obwodowego lub „nocycepcji”, (3) rdzenia kręgowego, (4) mózgu, (5) lokalizacji receptorów w barku i (6 ) anatomia nerwowa barku. Zastanawiamy się również, w jaki sposób czynniki te mogą przyczynić się do zmienności obrazu klinicznego, diagnozy i leczenia bólu barku. W ten sposób zamierzamy przedstawić przegląd elementów składowych systemu wykrywania bólu obwodowego i ośrodkowych mechanizmów przetwarzania bólu w bólu barku, które współdziałają w celu wywołania bólu klinicznego.
WPROWADZENIE: BARDZO KRÓTKA HISTORIA NAUKI BÓLU NIEZBĘDNEJ DLA KLINIKÓW
Ogólnie rzecz biorąc, natura bólu była przedmiotem wielu kontrowersji w ciągu ostatniego stulecia. W XVII-wiecznej teorii Kartezjusza17 sugerowano, że intensywność bólu jest bezpośrednio związana z rozmiarem uszkodzenia tkanki towarzyszącej i że ból jest przetwarzany jedną odrębną ścieżką. Wiele wcześniejszych teorii opierało się na tej tak zwanej „dualistycznej” filozofii kartezjańskiej, widząc ból jako konsekwencję stymulacji „specyficznego” obwodowego receptora bólu w mózgu. W XX wieku wywiązała się naukowa bitwa pomiędzy dwiema przeciwstawnymi teoriami, mianowicie teorią specyficzności i teorią wzorców. Kartezjańska „teoria specyficzności” postrzegała ból jako specyficzną, oddzielną modalność bodźców sensorycznych z własnym aparatem, podczas gdy „teoria wzorców” uważała, że ból jest wynikiem intensywnej stymulacji niespecyficznych receptorów.1 W 20 Wall i Melzack 2 teoria bramkowa bólu dostarczyła dowodów na istnienie modelu, w którym percepcja bólu była modulowana zarówno przez czuciowe sprzężenie zwrotne, jak i centralny układ nerwowy. Kolejnym ogromnym postępem w teorii bólu, mniej więcej w tym samym czasie, było odkrycie specyficznego sposobu działania opioidów.1965 Następnie ostatnie postępy w neuroobrazowaniu i medycynie molekularnej znacznie poszerzyły naszą ogólną wiedzę na temat bólu.
Więc jak to się ma do bólu barku?�Ból barku jest częstym problemem klinicznym, a dogłębne zrozumienie sposobu, w jaki ból jest przetwarzany przez organizm, jest niezbędne do najlepszego diagnozowania i leczenia bólu pacjenta. Postępy w naszej wiedzy na temat przetwarzania bólu obiecują wyjaśnić rozbieżność między patologią a odczuwaniem bólu, mogą również pomóc nam wyjaśnić, dlaczego niektórzy pacjenci nie reagują na określone terapie.
PODSTAWOWE BUDOWNICTWO BÓLU
Obwodowe receptory czuciowe: mechanoreceptor i „nocyceptor”
W ludzkim układzie mięśniowo-szkieletowym występuje wiele rodzajów obwodowych receptorów czuciowych. 5 Mogą być klasyfikowane na podstawie ich funkcji (jako mechanoreceptory, termoreceptory lub nocyceptory) lub morfologii (wolne zakończenia nerwowe lub różne typy receptorów zamkniętych w kapsułce).5 Różne typy receptorów można następnie dalej klasyfikować w oparciu o obecność niektórych markerów chemicznych. Istnieją znaczne nakładania się różnych klas funkcjonalnych receptora, na przykład:
Przetwarzanie bólu obwodowego: „Nocycepcja”
Uszkodzenie tkanki obejmuje różne mediatory zapalne uwalniane przez uszkodzone komórki, w tym bradykininę, histaminę, 5-hydroksytryptaminę, ATP, tlenek azotu i niektóre jony (K+ i H+). Aktywacja szlaku kwasu arachidonowego prowadzi do produkcji prostaglandyn, tromboksanów i leukotrienów. Cytokiny, w tym interleukiny i czynnik martwicy nowotworu a, oraz neurotrofiny, takie jak czynnik wzrostu nerwów (NGF), są również uwalniane i są ściśle zaangażowane w wywoływanie stanu zapalnego.15 Inne substancje, takie jak aminokwasy pobudzające (glutaminian) i opioidy ( endotelina-1) była również zaangażowana w ostrą odpowiedź zapalną.16 17 Niektóre z tych czynników mogą bezpośrednio aktywować nocyceptory, podczas gdy inne powodują rekrutację innych komórek, które następnie uwalniają kolejne czynniki ułatwiające.18 Ten lokalny proces skutkuje zwiększoną reaktywnością. neuronów nocyceptywnych do ich normalnego sygnału wejściowego i/lub rekrutacja odpowiedzi na sygnały normalnie podprogowe jest określana jako „uczulenie obwodowe”. Rysunek 1 podsumowuje niektóre kluczowe mechanizmy.
NGF i receptor członu 1 kanału kationowego podrodziny V podrodziny receptora przejściowego potencjalnego receptora (TRPV1) mają symbiotyczny związek, jeśli chodzi o zapalenie i uczulenie na receptory nocyceptorowe. Cytokiny wytwarzane w tkance w stanie zapalnym powodują wzrost produkcji NGF.19 NGF stymuluje uwalnianie histaminy i serotoniny (5-HT3) przez komórki tuczne, a także uwrażliwia nocyceptory, prawdopodobnie zmieniając właściwości A? włókna tak, że większa część staje się nocyceptywna. Receptor TRPV1 jest obecny w subpopulacji pierwotnych włókien doprowadzających i jest aktywowany przez kapsaicynę, ciepło i protony. Receptor TRPV1 jest syntetyzowany w ciele komórki włókna aferentnego i jest transportowany zarówno do terminali obwodowych, jak i centralnych, gdzie przyczynia się do wrażliwości neuronów aferentnych nocyceptywnych. Zapalenie powoduje obwodowe wytwarzanie NGF, który następnie wiąże się z receptorem kinazy tyrozynowej typu 1 na końcach nocyceptorów, NGF jest następnie transportowany do ciała komórki, gdzie prowadzi do regulacji w górę transkrypcji TRPV1, a w konsekwencji do zwiększonej wrażliwości na receptory nocyceptorowe.19 20 NGF i inne mediatory zapalne również uwrażliwiają TRPV1 poprzez różnorodną gamę drugorzędowych szlaków przekaźnikowych. Uważa się, że wiele innych receptorów, w tym receptory cholinergiczne, receptory kwasu y-aminomasłowego (GABA) i receptory somatostatyny, są również zaangażowane w wrażliwość obwodowych receptorów nocyceptorowych.
Duża liczba mediatorów zapalnych jest szczególnie związana z bólem barku i chorobą stożka rotatorów.21 Podczas gdy niektóre mediatory chemiczne bezpośrednio aktywują nocyceptory, większość z nich prowadzi do zmian w samym neuronie czuciowym, a nie bezpośrednio go aktywuje. Zmiany te mogą być zależne od wczesnej potranslacyjnej lub opóźnionej transkrypcji. Przykładami tych pierwszych są zmiany w receptorze TRPV25 lub w kanałach jonowych bramkowanych napięciem, wynikające z fosforylacji białek związanych z błoną. Przykłady tych ostatnich obejmują indukowany przez NGF wzrost wytwarzania kanału TRV1 i indukowaną wapniem aktywację wewnątrzkomórkowych czynników transkrypcyjnych.
Molekularne mechanizmy nocycepcji
Uczucie bólu ostrzega nas o prawdziwym lub zbliżającym się urazie i wyzwala odpowiednie reakcje ochronne. Niestety ból często przeżywa swoją przydatność jako system ostrzegawczy i zamiast tego staje się chroniczny i wyniszczający. To przejście do fazy przewlekłej obejmuje zmiany w rdzeniu kręgowym i mózgu, ale istnieje również niezwykła modulacja, w której inicjowane są komunikaty bólowe – na poziomie pierwotnego neuronu czuciowego. Wysiłki zmierzające do ustalenia, w jaki sposób te neurony wykrywają bodźce wywołujące ból o charakterze termicznym, mechanicznym lub chemicznym, ujawniły nowe mechanizmy sygnalizacji i przybliżyły nas do zrozumienia zdarzeń molekularnych, które ułatwiają przejście od bólu ostrego do uporczywego.
Neurochemia nocyceptorów
Glutaminian jest głównym neuroprzekaźnikiem pobudzającym we wszystkich nocyceptorach. Badania histochemiczne dorosłych DRG ujawniły jednak dwie szerokie klasy niezmielinizowanego włókna C.
Przetworniki chemiczne, aby pogorszyć ból
Jak opisano powyżej, uraz zwiększa nasze odczuwanie bólu poprzez zwiększenie wrażliwości nocyceptorów na bodźce zarówno termiczne, jak i mechaniczne. Zjawisko to wynika po części z wytwarzania i uwalniania mediatorów chemicznych z pierwotnego terminala czuciowego oraz z komórek nienerwowych (np. fibroblastów, komórek tucznych, neutrofili i płytek krwi) w środowisku36 (ryc. 3). Niektóre składniki zupy zapalnej (na przykład protony, ATP, serotonina lub lipidy) mogą bezpośrednio zmieniać pobudliwość neuronów poprzez interakcję z kanałami jonowymi na powierzchni nocyceptora, podczas gdy inne (na przykład bradykinina i NGF) wiążą się z receptorami metabotropowymi i pośredniczą w ich skutkach poprzez kaskady sygnalizacyjne drugiego posłańca11. Poczyniono znaczne postępy w zrozumieniu podstaw biochemicznych takich mechanizmów modulujących.
Protony zewnątrzkomórkowe i kwasica tkankowa
Miejscowa kwasica tkankowa jest charakterystyczną fizjologiczną reakcją na uraz, a stopień związanego z nią bólu lub dyskomfortu jest dobrze skorelowany ze stopniem zakwaszenia37. Podawanie kwasu (pH 5) na skórę powoduje długotrwałe wyładowania w co najmniej jednej trzeciej nocyceptorów polimodalnych, które unerwiają pole recepcyjne 20.
Komórkowe i molekularne mechanizmy bólu
Abstrakcyjny
Układ nerwowy wykrywa i interpretuje szeroką gamę bodźców termicznych i mechanicznych, a także środowiskowe i endogenne chemiczne czynniki drażniące. Gdy są intensywne, bodźce te generują ostry ból, aw sytuacji trwałego urazu, zarówno obwodowe, jak i ośrodkowe elementy układu nerwowego szlaku przekazywania bólu wykazują ogromną plastyczność, wzmacniając sygnały bólowe i wywołując nadwrażliwość. Kiedy plastyczność ułatwia odruchy ochronne, może to być korzystne, ale gdy zmiany utrzymują się, może wystąpić przewlekły ból. Badania genetyczne, elektrofizjologiczne i farmakologiczne wyjaśniają mechanizmy molekularne, które leżą u podstaw wykrywania, kodowania i modulowania szkodliwych bodźców wywołujących ból.
Wstęp: ostry kontra uporczywy ból
Rycina 5. Uczulenie rdzenia kręgowego (ośrodkowe)
Uczulenie za pośrednictwem receptora glutaminianu/NMDA.�Po intensywnej stymulacji lub uporczywym urazie, aktywowane C i A? nocyceptory uwalniają różne neuroprzekaźniki, w tym dlutaminian, substancję P, peptyd związany z genem kalcytoniny (CGRP) i ATP, do neuronów wyjściowych w blaszce I powierzchownego rogu grzbietowego (czerwony). W konsekwencji normalnie ciche receptory glutaminianu NMDA zlokalizowane w neuronie postsynaptycznym mogą teraz sygnalizować, zwiększać poziom wapnia wewnątrzkomórkowego i aktywować wiele ścieżek sygnałowych zależnych od wapnia i wtórnych przekaźników, w tym kinazę białkową aktywowaną mitogenem (MAPK), kinazę białkową C (PKC) , kinaza białkowa A (PKA) i Src. Ta kaskada zdarzeń zwiększy pobudliwość neuronu wyjściowego i ułatwi przekazywanie wiadomości bólowych do mózgu.
Odhamowanie.„W normalnych warunkach interneurony hamujące (niebieskie) w sposób ciągły uwalniają GABA i/lub glicynę (Gly), aby zmniejszyć pobudliwość neuronów wyjściowych blaszki I i modulować przenoszenie bólu (ton hamujący). Jednak w sytuacji urazu hamowanie to może zostać utracone, co prowadzi do przeczulicy bólowej. Dodatkowo, odhamowanie może umożliwić nienocyceptywną mielinizację A? pierwotne aferenty angażujące obwody transmisji bólu tak, że normalnie nieszkodliwe bodźce są teraz odbierane jako bolesne. Dzieje się to po części poprzez odhamowanie pobudzającego PKC? wyrażanie interneuronów w wewnętrznej blaszce II.
Aktywacja mikrogleju.„Uszkodzenie nerwów obwodowych sprzyja uwalnianiu ATP i chemokiny fraktalkiny, które będą stymulować komórki mikrogleju. W szczególności aktywacja receptorów purynergicznych, CX3CR1 i Toll-podobnych na mikrogleju (fioletowy) powoduje uwolnienie neurotroficznego czynnika pochodzenia mózgowego (BDNF), który poprzez aktywację receptorów TrkB wyrażanych przez neurony wyprowadzające blaszkę I, sprzyja zwiększonej pobudliwości i nasilony ból w odpowiedzi zarówno na szkodliwą, jak i nieszkodliwą stymulację (tj. przeczulicę bólową i allodynię). Aktywowany mikroglej uwalnia również wiele cytokin, takich jak czynnik martwicy nowotworu? (TNF?), interleukina-1? i 6 (IL-la, IL-1) oraz inne czynniki, które przyczyniają się do uczulenia ośrodkowego.
Chemiczne środowisko zapalenia
Uczulenie obwodowe częściej wynika ze zmian związanych ze stanem zapalnym w środowisku chemicznym włókna nerwowego (McMahon i wsp., 2008). Tak więc uszkodzeniu tkanki często towarzyszy akumulacja endogennych czynników uwalnianych z aktywowanych nocyceptorów lub komórek nieneuronalnych, które znajdują się w uszkodzonym obszarze lub przenikają do niego (w tym komórki tuczne, bazofile, płytki krwi, makrofagi, neutrofile, komórki śródbłonka, keratynocyty i fibroblasty). Zbiorowo. czynniki te, określane mianem „zupy zapalnej”, reprezentują szeroki wachlarz cząsteczek sygnałowych, w tym neuroprzekaźniki, peptydy (substancja P, CGRP, bradykinina), eikozynoidy i pokrewne lipidy (prostaglandyny, tromboksany, leukotrieny, endokannabinoidy), neurotrofiny, cytokiny oraz chemokiny, a także proteazy zewnątrzkomórkowe i protony. Co godne uwagi, nocyceptory wyrażają jeden lub więcej receptorów powierzchniowych komórek zdolnych do rozpoznawania i odpowiadania na każdy z tych czynników prozapalnych lub proalgetycznych (Figura 4). Takie interakcje zwiększają pobudliwość włókna nerwowego, zwiększając w ten sposób jego wrażliwość na temperaturę lub dotyk.
Niewątpliwie najczęstsze podejście do zmniejszania bólu zapalnego polega na hamowaniu syntezy lub akumulacji składników zapalnej zupy. Najlepszym przykładem są niesteroidowe leki przeciwzapalne, takie jak aspiryna lub ibuprofen, które zmniejszają ból zapalny i przeczulicę bólową poprzez hamowanie cyklooksygenaz (Cox-1 i Cox-2) zaangażowanych w syntezę prostaglandyn. Drugie podejście polega na zablokowaniu działania czynników zapalnych w nocyceptorze. W tym miejscu przedstawiamy przykłady, które zapewniają nowy wgląd w komórkowe mechanizmy uczulenia obwodowego lub które stanowią podstawę nowych strategii terapeutycznych w leczeniu bólu zapalnego.
NGF jest prawdopodobnie najlepiej znany ze swojej roli jako czynnika neurotroficznego wymaganego do przeżycia i rozwoju neuronów czuciowych podczas embriogenezy, ale u dorosłych NGF jest również wytwarzany w warunkach uszkodzenia tkanki i stanowi ważny składnik zupy zapalnej (Ritner et al. al., 2009). Wśród wielu celów komórkowych, NGF działa bezpośrednio na nocyceptory peptydergicznych włókien C, które wyrażają kinazę tyrozynową receptora NGF o wysokim powinowactwie, TrkA, jak również receptor neurotrofiny o niskim powinowactwie, p75 (Chao, 2003; Snider i McMahon, 1998). NGF wytwarza głęboką nadwrażliwość na ciepło i bodźce mechaniczne dzięki dwóm czasowo odrębnym mechanizmom. Początkowo oddziaływanie NGF-TrkA aktywuje dalsze szlaki sygnałowe, w tym fosfolipazę C (PLC), kinazę białkową aktywowaną mitogenem (MAPK) i kinazę 3-fosfoinozytydu (PI3K). Powoduje to funkcjonalne wzmocnienie białek docelowych na obwodowym końcu nocyceptora, w szczególności TRPV1, prowadząc do szybkiej zmiany wrażliwości komórkowej i behawioralnej na ciepło (Chuang i wsp., 2001).
Niezależnie od ich mechanizmów pronocyceptywnych, zakłócanie sygnalizacji neurotrofin lub cytokin stało się główną strategią kontrolowania choroby zapalnej lub wynikającego z niej bólu. Główne podejście polega na blokowaniu NGF lub TNF-? działanie z neutralizującym przeciwciałem. W przypadku TNF-a, okazało się to niezwykle skuteczne w leczeniu licznych chorób autoimmunologicznych, w tym reumatoidalnego zapalenia stawów, prowadząc do dramatycznego zmniejszenia zarówno zniszczenia tkanki, jak i towarzyszącej hiperalgezji (Atzeni i wsp., 2005). Ponieważ główne działania NGF na receptory nocyceptorów dorosłych występują w warunkach zapalenia, zaletą tego podejścia jest zmniejszenie przeczulicy bólowej bez wpływu na normalne postrzeganie bólu. Rzeczywiście, przeciwciała anty-NGF są obecnie w badaniach klinicznych w leczeniu zespołów bólu zapalnego (Hefti i wsp., 2006).
Uczulenie za pośrednictwem receptora glutaminianu/NMDA
Ostry ból jest sygnalizowany uwalnianiem glutaminianu z centralnych końcówek nocyceptorów, generując pobudzające prądy postsynaptyczne (EPSC) w neuronach rogu grzbietowego drugiego rzędu. Dzieje się to głównie poprzez aktywację postsynaptycznych podtypów AMPA i kainowych jonotropowych receptorów glutaminianu. Sumowanie podprogowych EPSC w neuronie postsynaptycznym w końcu spowoduje odpalenie potencjału czynnościowego i przekazanie wiadomości o bólu do neuronów wyższego rzędu.
Inne badania wskazują, że same zmiany w neuronie projekcyjnym przyczyniają się do procesu odhamowania. Na przykład, uszkodzenie nerwów obwodowych głęboko obniża poziom transportera K+-Cl- KCC2, który jest niezbędny do utrzymania normalnych gradientów K+ i Cl- przez błonę plazmatyczną (Coull i wsp., 2003). Regulacja w dół KCC2, która jest wyrażana w neuronach projekcyjnych blaszki I, powoduje przesunięcie gradientu Cl-, tak że aktywacja receptorów GABA-A depolaryzuje, zamiast hiperpolaryzuje neurony projekcyjne blaszki I. To z kolei zwiększyłoby pobudliwość i zwiększyłoby przenoszenie bólu. Rzeczywiście, blokada farmakologiczna lub regulacja w dół KCC2 za pośrednictwem siRNA u szczura indukuje mechaniczną allodynię.
Udostępnij Ebook
Źródła:
Dlaczego boli mnie ramię? Przegląd neuroanatomicznych i biochemicznych podstaw bólu barku
Benjamina Johna Floyda Deana, Stephena Edwarda Gwilyma i Andrew Jonathana Carra
Komórkowe i molekularne mechanizmy bólu
Allan I. Basbaum1, Diana M. Bautista2, Gregory Scherrer1 i David Julius3
1Katedra Anatomii, Uniwersytet Kalifornijski, San Francisco 94158
2 Wydział Biologii Molekularnej i Komórkowej, Uniwersytet Kalifornijski, Berkeley CA 94720 3 Wydział Fizjologii Uniwersytetu Kalifornijskiego, San Francisco 94158
Molekularne mechanizmy nocycepcji
David Julius* i Allan I. Basbaum�
*Katedra Farmakologii Komórkowej i Molekularnej oraz Wydziały Anatomii i Fizjologii oraz WM Keck Foundation Center for Integrative Neuroscience, University of California San Francisco, San Francisco, California 94143, USA (e-mail: julius@socrates.ucsf.edu)
Zapalenie neurogenne, lub NI, jest procesem fizjologicznym, w którym mediatory są uwalniane bezpośrednio z nerwów skórnych, aby rozpocząć odpowiedź zapalną. Powoduje to powstawanie miejscowych reakcji zapalnych, w tym rumienia, obrzęku, wzrostu temperatury, tkliwości i bólu. Drobne niezmielinizowane aferentne somatyczne włókna C, które reagują na stymulację mechaniczną i chemiczną o niskiej intensywności, są w dużej mierze odpowiedzialne za uwalnianie tych mediatorów zapalnych.
Po stymulacji te szlaki nerwowe w nerwach skórnych szybko uwalniają energetyczne neuropeptydy lub substancję P i peptyd związany z genem kalcytoniny (CGRP) do mikrośrodowiska, wywołując serię odpowiedzi zapalnych. Istnieje znacząca różnica w zapaleniu immunogennym, które jest pierwszą ochronną i naprawczą odpowiedzią układu odpornościowego, gdy patogen dostanie się do organizmu, podczas gdy zapalenie neurogenne obejmuje bezpośrednie połączenie między układem nerwowym a reakcjami zapalnymi. Chociaż zapalenie neurogenne i zapalenie immunologiczne mogą występować jednocześnie, nie są one klinicznie nie do odróżnienia. Celem poniższego artykułu jest omówienie mechanizmu zapalenia neurogennego oraz roli obwodowego układu nerwowego w obronie gospodarza i immunopatologii.
Zapalenie neurogenne – rola obwodowego układu nerwowego w obronie gospodarza i immunopatologii
Abstrakcyjny
Tradycyjnie uważa się, że obwodowy układ nerwowy i odpornościowy pełnią odrębne funkcje. Ta linia jest jednak coraz bardziej zacierana przez nowe wglądy w neurogenne zapalenie. Neurony nocyceptorowe posiadają wiele takich samych ścieżek molekularnego rozpoznawania niebezpieczeństw jak komórki odpornościowe, aw odpowiedzi na niebezpieczeństwo obwodowy układ nerwowy komunikuje się bezpośrednio z układem odpornościowym, tworząc zintegrowany mechanizm ochronny. Gęsta sieć unerwienia włókien czuciowych i autonomicznych w tkankach obwodowych oraz duża szybkość transdukcji neuronalnej pozwala na szybką lokalną i ogólnoustrojową neurogenną modulację odporności. Wydaje się, że neurony obwodowe również odgrywają istotną rolę w dysfunkcji układu odpornościowego w chorobach autoimmunologicznych i alergicznych. Dlatego zrozumienie skoordynowanej interakcji neuronów obwodowych z komórkami układu odpornościowego może przyczynić się do rozwoju metod terapeutycznych w celu zwiększenia obrony gospodarza i tłumienia immunopatologii.
Wprowadzenie
Dwa tysiące lat temu Celsus zdefiniował stan zapalny jako obejmujący cztery główne objawy: ból (ból), kalor (ciepło), rubor (zaczerwienienie) i guz (obrzęk). zapalenie. Jednak od tego czasu ból był uważany głównie za objaw, a nie jako uczestnik powstawania stanu zapalnego. Z tej perspektywy pokazujemy, że obwodowy układ nerwowy odgrywa bezpośrednią i aktywną rolę w modulowaniu odporności wrodzonej i nabytej, tak że układy odpornościowy i nerwowy mogą mieć wspólną zintegrowaną funkcję ochronną w obronie gospodarza i odpowiedzi na uszkodzenie tkanki, co jest skomplikowane. interakcje, które również mogą prowadzić do patologii w chorobach alergicznych i autoimmunologicznych.
Przeżycie organizmów jest krytycznie zależne od zdolności do obrony przed potencjalnymi szkodami spowodowanymi uszkodzeniem tkanki i infekcją. Obrona gospodarza obejmuje zarówno zachowanie unikające, aby usunąć kontakt z niebezpiecznym (szkodliwym) środowiskiem (funkcja nerwowa), jak i aktywną neutralizację patogenów (funkcja immunologiczna). Tradycyjnie uważa się, że rola układu odpornościowego w zwalczaniu czynników zakaźnych i naprawie uszkodzeń tkanek jest całkiem odmienna od roli układu nerwowego, który przetwarza szkodliwe sygnały środowiskowe i wewnętrzne na aktywność elektryczną, aby wywoływać odczucia i odruchy (ryc. 1). Proponujemy, aby te dwa systemy były w rzeczywistości składnikami zunifikowanego mechanizmu obronnego. Somatosensoryczny układ nerwowy jest idealnie przystosowany do wykrywania niebezpieczeństwa. Po pierwsze, wszystkie tkanki silnie narażone na działanie środowiska zewnętrznego, takie jak nabłonkowe powierzchnie skóry, płuca, układ moczowy i pokarmowy, są gęsto unerwione przez nocyceptory, czyli włókna czuciowe o wysokim progu bólowym. Po drugie, transdukcja szkodliwych bodźców zewnętrznych jest niemal natychmiastowa, o rząd wielkości szybsza niż mobilizacja wrodzonego układu odpornościowego, a zatem może być „pierwszą odpowiedzią” w obronie gospodarza.
Rysunek 1: Szkodliwe bodźce, ścieżki rozpoznawania drobnoustrojów i stanów zapalnych wywołują aktywację obwodowego układu nerwowego. Neurony czuciowe posiadają kilka sposobów wykrywania obecności szkodliwych/szkodliwych bodźców. 1) Receptory sygnałów niebezpiecznych, w tym kanały TRP, kanały P2X i receptory związane z niebezpieczeństwem molekularnym (DAMP) rozpoznają egzogenne sygnały z otoczenia (np. ciepło, kwasowość, chemikalia) lub endogenne sygnały niebezpieczeństwa uwalniane podczas urazu/uszkodzenia tkanki (np. ATP, kwas moczowy, hydroksynonenale). 2) Receptory rozpoznawania wzorców (PRR), takie jak receptory Toll-podobne (TLR) i receptory typu Nod (NLR) rozpoznają wzorce molekularne związane z patogenem (PAMP) wydzielane przez atakujące bakterie lub wirusy podczas infekcji. 3) Receptory cytokinowe rozpoznają czynniki wydzielane przez komórki odpornościowe (np. IL-1beta, TNF-alfa, NGF), które aktywują kinazy mapowe i inne mechanizmy sygnalizacyjne w celu zwiększenia pobudliwości błony.
Oprócz ortodromicznych sygnałów wejściowych do rdzenia kręgowego i mózgu z obwodu, potencjały czynnościowe w neuronach nocyceptorowych mogą być również przekazywane antydromicznie w punktach rozgałęzień z powrotem do obwodu, czyli odruchu aksonu. Te, wraz z utrzymującymi się lokalnymi depolaryzacjami, prowadzą do szybkiego i lokalnego uwalniania mediatorów nerwowych zarówno z aksonów obwodowych, jak i końcówek (ryc. 2) 1. Klasyczne eksperymenty Goltza (w 1874) i Bayliss (w 1901) wykazały, że elektrycznie stymulujące korzenie grzbietowe indukuje rozszerzenie naczyń skóry, co doprowadziło do koncepcji „zapalenia neurogennego”, niezależnego od tego wytwarzanego przez układ odpornościowy (ryc. 3).
Rysunek 2: Czynniki neuronalne uwalniane z nocyceptorowych neuronów czuciowych bezpośrednio kierują chemotaksją leukocytów, hemodynamiką naczyń i odpowiedzią immunologiczną. Gdy szkodliwe bodźce aktywują sygnały aferentne w nerwach czuciowych, generowane są antydromowe odruchy aksonów, które indukują uwalnianie neuropeptydów na obwodowych zakończeniach neuronów. Te mediatory molekularne mają kilka działań zapalnych: 1) Chemotaksja i aktywacja neutrofili, makrofagów i limfocytów w miejscu uszkodzenia oraz degranulacja komórek tucznych. 2) Sygnalizacja do komórek śródbłonka naczyniowego w celu zwiększenia przepływu krwi, przecieku naczyniowego i obrzęku. Pozwala to również na łatwiejszą rekrutację leukocytów zapalnych. 3) Przygotowywanie komórek dendrytycznych w celu napędzania dalszego różnicowania limfocytów T pomocniczych do podtypów Th2 lub Th17.
Rysunek 3: Kalendarium postępów w zrozumieniu neurogennych aspektów zapalenia od Celsusa do dnia dzisiejszego.
W zapaleniu neurogennym pośredniczy uwalnianie neuropeptydów peptydu związanego z genem kalcytoniny (CGRP) i substancji P (SP) z nocyceptorów, które działają bezpośrednio na komórki śródbłonka naczyń i mięśni gładkich 2. CGRP wywołuje efekt rozszerzenia naczyń 5, 2, podczas gdy SP zwiększa przepuszczalność naczyń włosowatych, prowadząc do wynaczynienia osocza i obrzęku 3, 4, przyczyniając się do powstawania szorstkości, kaloryczności i guza Celsusa. Jednak nocyceptory uwalniają wiele dodatkowych neuropeptydów (baza danych online: www.neuropeptydy.nl/), w tym adrenomedulina, neurokininy A i B, wazoaktywny peptyd jelitowy (VIP), neuropeptyd (NPY) i peptyd uwalniający gastrynę (GRP), a także inne mediatory molekularne, takie jak glutaminian, tlenek azotu (NO) i cytokiny, takie jak eotaksyna 6.
Teraz doceniamy, że mediatory uwalniane z neuronów czuciowych na obwodzie nie tylko działają na układ naczyniowy, ale także bezpośrednio przyciągają i aktywują komórki odporności wrodzonej (komórki tuczne, komórki dendrytyczne) i komórki odporności nabytej (limfocyty T) 7�12. Przypuszczamy, że w ostrym stanie uszkodzenia tkanki neurogenne zapalenie ma działanie ochronne, ułatwiając fizjologiczne gojenie ran i obronę immunologiczną przeciwko patogenom poprzez aktywację i rekrutację komórek odpornościowych. Jednak taka komunikacja neuroimmunologiczna prawdopodobnie odgrywa również główną rolę w patofizjologii chorób alergicznych i autoimmunologicznych poprzez wzmacnianie patologicznych lub nieprzystosowawczych odpowiedzi immunologicznych. Na przykład na modelach zwierzęcych reumatoidalnego zapalenia stawów Levine i współpracownicy wykazali, że odnerwienie stawu prowadzi do uderzającego osłabienia zapalenia, które jest zależne od ekspresji neuronalnej substancji P 13, 14. W ostatnich badaniach nad alergicznym zapaleniem dróg oddechowych, zapaleniem okrężnicy i łuszczyca, pierwotne neurony czuciowe odgrywają kluczową rolę w inicjowaniu i zwiększaniu aktywacji odporności wrodzonej i nabytej 15.
Proponujemy zatem, aby obwodowy układ nerwowy odgrywał nie tylko bierną rolę w obronie gospodarza (wykrywanie szkodliwych bodźców i inicjowanie zachowań unikających), ale także aktywną rolę we współpracy z układem odpornościowym w modulowaniu odpowiedzi i zwalczaniu szkodliwych bodźce, rolę, którą można odwrócić, aby przyczynić się do choroby.
Wspólne ścieżki rozpoznawania zagrożeń w obwodowych układach nerwowych i wrodzonej odporności
Obwodowe neurony czuciowe są przystosowane do rozpoznawania zagrożeń dla organizmu dzięki wrażliwości na intensywne bodźce mechaniczne, termiczne i drażniące chemiczne (ryc. 1). Kanały jonowe przejściowego potencjału receptora (TRP) są najszerzej zbadanymi molekularnymi mediatorami nocycepcji, prowadzącymi nieselektywne wnikanie kationów po aktywacji przez różne szkodliwe bodźce. TRPV1 jest aktywowany przez wysokie temperatury, niskie pH i kapsaicynę, walinoidowy drażniący składnik papryczek chili 18. TRPA1 pośredniczy w wykrywaniu reaktywnych substancji chemicznych, w tym drażniących środowiskowych, takich jak gaz łzawiący i izotiocyjaniany przemysłowe 19, ale co ważniejsze, jest również aktywowany w tkance uszkodzenie przez endogenne sygnały molekularne, w tym 4-hydroksynonenal i prostaglandyny 20, 21.
Co ciekawe, neurony czuciowe mają wiele tych samych patogennych i niebezpiecznych szlaków receptorowych rozpoznawania molekularnego, co komórki wrodzonej odporności, co umożliwia im również wykrywanie patogenów (ryc. 1). W układzie odpornościowym patogeny drobnoustrojowe są wykrywane przez receptory rozpoznawania wzorców kodowanych w linii zarodkowej (PRR), które rozpoznają szeroko konserwatywne wzorce molekularne związane z patogenami egzogennymi (PAMP). Pierwszymi zidentyfikowanymi PRR byli członkowie rodziny receptorów Toll-podobnych (TLR), które wiążą się z drożdżami, składnikami ściany komórkowej pochodzenia bakteryjnego i wirusowym RNA 22. Po aktywacji PRR włączane są dalsze szlaki sygnałowe, które indukują wytwarzanie i aktywację cytokin odporności adaptacyjnej. Oprócz TLR, wrodzone komórki odpornościowe są aktywowane podczas uszkodzenia tkanki przez endogenne sygnały zagrożenia, znane również jako wzorce molekularne związane z uszkodzeniem (DAMP) lub alarminy 23, 24. Te sygnały zagrożenia obejmują uwalniane HMGB1, kwas moczowy i białka szoku cieplnego przez umierające komórki podczas martwicy, aktywację komórek odpornościowych podczas niezakaźnych odpowiedzi zapalnych.
PRR, w tym TLR 3, 4, 7 i 9, są wyrażane przez neurony nocyceptorowe, a stymulacja przez ligandy TLR prowadzi do indukcji prądów wewnętrznych i uwrażliwienia nocyceptorów na inne bodźce bólowe 25. Ponadto aktywacja neuronów czuciowych przez ligand TLR27 imikwimod prowadzi do aktywacji szlaku czuciowego specyficznego dla świądu7. Wyniki te wskazują, że ból i swędzenie związane z infekcją mogą być częściowo spowodowane bezpośrednią aktywacją neuronów przez czynniki pochodzące od patogenów, co z kolei aktywować komórki odpornościowe poprzez obwodowe uwalnianie neuronalnych cząsteczek sygnalizacyjnych.
Głównym DAMP/alarminem uwalnianym podczas uszkodzenia komórki jest ATP, który jest rozpoznawany przez receptory purynergiczne zarówno w neuronach nocyceptorowych, jak i komórkach odpornościowych28. Receptory purynergiczne składają się z dwóch rodzin: receptorów P30X, kanałów kationowych bramkowanych ligandami i receptorów P2Y, receptorów sprzężonych z białkiem G. W neuronach nocyceptorowych rozpoznawanie ATP następuje przez P2X2, co prowadzi do szybkiego zagęszczania prądów kationowych i bólu 3, 28 (ryc. 30), podczas gdy receptory P1Y przyczyniają się do aktywacji nocyceptorów poprzez uwrażliwienie TRP i kanałów sodowych bramkowanych napięciem. W makrofagach wiązanie ATP z receptorami P2X2 prowadzi do hiperpolaryzacji i dalszej aktywacji inflamasomu, kompleksu molekularnego ważnego w wytwarzaniu IL-7beta i IL-1. Dlatego ATP jest silnym sygnałem niebezpieczeństwa, który aktywuje zarówno neurony obwodowe, jak i wrodzone. odporność podczas urazu, a niektóre dowody sugerują nawet, że neurony eksprymują części maszynerii molekularnej inflamasomu 18.
Drugą stroną sygnałów zagrożenia w nocyceptorach jest rola kanałów TRP w aktywacji komórek odpornościowych. TRPV2, homolog TRPV1 aktywowany szkodliwym ciepłem, jest eksprymowany na wysokim poziomie we wrodzonych komórkach odpornościowych 32. Genetyczna ablacja TRPV2 prowadziła do defektów fagocytozy makrofagów i usuwania infekcji bakteryjnych 32. Komórki tuczne również wyrażają kanały TRPV, które mogą bezpośrednio pośredniczyć ich degranulacja 33. Pozostaje do ustalenia, czy endogenne sygnały niebezpieczeństwa aktywują komórki odpornościowe w podobny sposób jak nocyceptory.
Kluczowym środkiem komunikacji między komórkami odpornościowymi a neuronami nocyceptorowymi są cytokiny. Po aktywacji receptorów cytokin, szlaki transdukcji sygnału są aktywowane w neuronach czuciowych, prowadząc do dalszej fosforylacji białek błonowych, w tym TRP i kanałów bramkowanych napięciem (ryc. 1). Wynikające z tego uczulenie nocyceptorów oznacza, że normalnie nieszkodliwe bodźce mechaniczne i cieplne mogą teraz aktywować nocyceptory. Interleukina 1 beta i TNF-alfa to dwie ważne cytokiny uwalniane przez wrodzone komórki odpornościowe podczas zapalenia. IL-1beta i TNF-alfa są bezpośrednio wyczuwane przez nocyceptory, które wyrażają pokrewne receptory, indukują aktywację kinaz mapy p38, prowadząc do zwiększonej pobudliwości błony 34. Czynnik wzrostu nerwów (NGF) i prostaglandyna E(36) są również głównymi mediatorami stanu zapalnego uwalnianymi z komórek odpornościowych, które działają bezpośrednio na obwodowe neurony czuciowe, powodując uczulenie. Ważnym efektem uczulenia nocyceptorów przez czynniki immunologiczne jest zwiększone uwalnianie neuropeptydów na zakończeniach obwodowych, które dodatkowo aktywują komórki odpornościowe, indukując w ten sposób pętlę dodatniego sprzężenia zwrotnego, która napędza i ułatwia stan zapalny.
Sensoryczna kontrola układu nerwowego odporności wrodzonej i adaptacyjnej
We wczesnych fazach zapalenia neurony czuciowe wysyłają sygnały do komórek tucznych rezydujących w tkankach i komórek dendrytycznych, które są wrodzonymi komórkami odpornościowymi ważnymi w inicjowaniu odpowiedzi immunologicznej (ryc. 2). Badania anatomiczne wykazały bezpośrednie przyłożenie końcówek do komórek tucznych, jak również do komórek dendrytycznych, a neuropeptydy uwalniane z nocyceptorów mogą indukować degranulację lub produkcję cytokin w tych komórkach 7, 9, 37. Ta interakcja odgrywa ważną rolę w alergicznych drogach oddechowych zapalenie i zapalenie skóry 10.
Podczas efektorowej fazy zapalenia komórki odpornościowe muszą znaleźć drogę do konkretnego miejsca uszkodzenia. Wiele mediatorów uwalnianych z neuronów czuciowych, neuropeptydów, chemokin i glutaminianu działa chemotaktycznie na neutrofile, eozynofile, makrofagi i limfocyty T oraz zwiększa adhezję śródbłonka, co ułatwia zasiedlanie komórek układu odpornościowego 6, 38 (ryc. 41). Ponadto niektóre dowody sugerują, że neurony mogą bezpośrednio uczestniczyć w fazie efektorowej, ponieważ same neuropeptydy mogą mieć bezpośrednie funkcje przeciwdrobnoustrojowe 2.
Cząsteczki sygnałowe pochodzenia neuronowego mogą również kierować rodzajem zapalenia, przyczyniając się do różnicowania lub specyfikacji różnych typów adaptacyjnych limfocytów T układu odpornościowego. Antygen jest fagocytowany i przetwarzany przez wrodzone komórki odpornościowe, które następnie migrują do najbliższego węzła chłonnego i prezentują peptyd antygenowy natywnym limfocytom T. W zależności od typu antygenu, cząsteczek kostymulujących na wrodzonej komórce odpornościowej i kombinacji określonych cytokin, natywne limfocyty T dojrzewają do określonych podtypów, które najlepiej służą wysiłkowi zapalnemu w celu usunięcia patogennego bodźca. Limfocyty T CD4 lub limfocyty T pomocnicze (Th) można podzielić na cztery główne grupy: Th1, Th2, Th17 i limfocyty T regulatorowe (Treg). Komórki Th1 biorą udział głównie w regulacji odpowiedzi immunologicznych na wewnątrzkomórkowe mikroorganizmy i narządowe choroby autoimmunologiczne; Th2 mają kluczowe znaczenie dla odporności przeciwko patogenom zewnątrzkomórkowym, takim jak robaki pasożytnicze, i są odpowiedzialne za alergiczne choroby zapalne; Komórki Th17 odgrywają kluczową rolę w ochronie przed wyzwaniami drobnoustrojów, takimi jak bakterie i grzyby pozakomórkowe; Komórki Treg biorą udział w utrzymywaniu samotolerancji i regulowaniu odpowiedzi immunologicznych. Wydaje się, że na ten proces dojrzewania komórek T duży wpływ mają mediatory neuronów czuciowych. Neuropeptydy, takie jak CGRP i VIP, mogą skłaniać komórki dendrytyczne do odporności typu Th2 i zmniejszać odporność typu Th1 poprzez promowanie wytwarzania niektórych cytokin i hamowanie innych, a także poprzez zmniejszanie lub zwiększanie migracji komórek dendrytycznych do lokalnych węzłów chłonnych8 , 10, 43. Neurony czuciowe również znacząco przyczyniają się do alergicznego (głównie napędzanego przez Th2) zapalenia 17. Oprócz regulacji komórek Th1 i Th2, inne neuropeptydy, takie jak SP i Hemokinina-1, mogą bardziej kierować odpowiedź zapalną w kierunku Th17 lub Treg 44, 45, co oznacza, że neurony mogą również brać udział w regulowaniu procesu zapalnego. W immunopatologiach, takich jak zapalenie okrężnicy i łuszczyca, blokada mediatorów neuronalnych, takich jak substancja P, może znacząco tłumić uszkodzenia limfocytów T i immunologiczne 15, chociaż antagonizowanie jednego mediatora może samo w sobie mieć jedynie ograniczony wpływ na zapalenie neurogenne.
Biorąc pod uwagę, że cząsteczki sygnałowe uwalniane z obwodowych włókien nerwów czuciowych regulują nie tylko małe naczynia krwionośne, ale także chemotaksję, naprowadzanie, dojrzewanie i aktywację komórek odpornościowych, staje się jasne, że interakcje neuroimmunologiczne są znacznie bardziej skomplikowane niż wcześniej sądzono (ryc. 2). Co więcej, można sobie wyobrazić, że to nie pojedyncze mediatory nerwowe, ale raczej specyficzne kombinacje cząsteczek sygnałowych uwalnianych z nocyceptorów wpływają na różne etapy i typy odpowiedzi immunologicznych.
Autonomiczna kontrola odruchów odporności
Ważną rolę odgrywa również „obwód odruchowy” cholinergicznego autonomicznego układu nerwowego w regulacji obwodowych odpowiedzi immunologicznych 46. Nerw błędny jest głównym nerwem przywspółczulnym łączącym pień mózgu z narządami trzewnymi. Praca Kevina Tracey i innych wskazuje na silne uogólnione reakcje przeciwzapalne we wstrząsie septycznym i endotoksemii, wywołane przez eferentną aktywność nerwu błędnego, prowadzącą do supresji makrofagów obwodowych47. Błąd błędny aktywuje neurony obwodowego adrenergicznego zwoju trzewnego unerwiające śledzionę, co prowadzi do dalszego uwalniania acetylocholiny, która wiąże się z receptorami nikotynowymi alfa-49 na makrofagach w śledzionie i przewodzie pokarmowym. Indukuje to aktywację szlaku sygnałowego JAK7/STAT2 SOCS3, który silnie hamuje transkrypcję TNF-alfa 3. Adrenergiczny zwój trzewny komunikuje się również bezpośrednio z podzbiorem limfocytów T pamięci wytwarzających acetylocholinę, które hamują zapalne makrofagi 47.
Niezmienne limfocyty T naturalnego zabójcy (iNKT) to wyspecjalizowany podzbiór limfocytów T, który rozpoznaje lipidy drobnoustrojów w kontekście CD1d zamiast antygenów peptydowych. Komórki NKT stanowią kluczową populację limfocytów zaangażowaną w zwalczanie patogenów zakaźnych i regulację odporności ogólnoustrojowej. Komórki NKT przebywają i przemieszczają się głównie przez układ naczyniowy i zatoki śledziony i wątroby. Współczulne nerwy beta-adrenergiczne w wątrobie bezpośrednio sygnalizują modulację aktywności komórek NKT 50. Na przykład podczas mysiego modelu udaru (MCAO), mobilność komórek NKT wątroby była wyraźnie stłumiona, co zostało odwrócone przez odnerwienie współczulne lub antagonistów beta-adrenergicznych. Co więcej, ta immunosupresyjna aktywność neuronów noradrenergicznych na komórkach NKT doprowadziła do wzrostu infekcji ogólnoustrojowej i uszkodzenia płuc. Dlatego sygnały odprowadzające z neuronów autonomicznych mogą pośredniczyć w silnej immunosupresji.
Wgląd doktora Alexa Jimeneza
Zapalenie neurogenne to lokalna reakcja zapalna generowana przez układ nerwowy. Uważa się, że odgrywa fundamentalną rolę w patogenezie różnych problemów zdrowotnych, w tym migreny, łuszczycy, astmy, fibromialgii, egzemy, trądziku różowatego, dystonii i nadwrażliwości chemicznej. Chociaż zapalenie neurogenne związane z obwodowym układem nerwowym zostało szeroko zbadane, koncepcja zapalenia neurogennego w obrębie ośrodkowego układu nerwowego nadal wymaga dalszych badań. Jednak według kilku badań naukowych uważa się, że niedobór magnezu jest główną przyczyną zapalenia neurogennego. Poniższy artykuł przedstawia przegląd mechanizmów neurogennego zapalenia w układzie nerwowym, który może pomóc pracownikom służby zdrowia określić najlepsze podejście do leczenia różnych problemów zdrowotnych związanych z układem nerwowym.
wnioski
Jaka jest odpowiednia rola somatosensorycznego i autonomicznego układu nerwowego w regulacji stanu zapalnego i układu odpornościowego (ryc. 4)? Aktywacja nocyceptorów prowadzi do lokalnych odruchów aksonowych, które lokalnie rekrutują i aktywują komórki odpornościowe, a zatem są głównie prozapalne i ograniczone przestrzennie. W przeciwieństwie do tego, stymulacja autonomiczna prowadzi do ogólnoustrojowej immunosupresji poprzez wpływ na pule komórek odpornościowych w wątrobie i śledzionie. Słabo poznane są aferentne mechanizmy sygnalizacji obwodowej prowadzące do wyzwolenia immunosupresyjnego układu odruchu cholinergicznego układu nerwowego błędnego. Jednak 80% włókien nerwu błędnego to pierwotne aferentne włókna czuciowe, a zatem sygnały z wnętrzności, z których wiele potencjalnie jest napędzanych przez komórki układu odpornościowego, może prowadzić do aktywacji interneuronów w pniu mózgu, a przez nie do wyjścia we włóknach eferentnych nerwu błędnego 90.
Rysunek 4: Sensoryczny i autonomiczny układ nerwowy modulują odpowiednio lokalną i ogólnoustrojową odpowiedź immunologiczną. Nocyceptory unerwiające powierzchnie nabłonka (np. skórę i płuca) wywołują zlokalizowane odpowiedzi zapalne, aktywując komórki tuczne i komórki dendrytyczne. W alergicznym zapaleniu dróg oddechowych, zapaleniu skóry i reumatoidalnym zapaleniu stawów neurony nocyceptorowe odgrywają rolę w wywoływaniu stanu zapalnego. Natomiast obwody autonomiczne unerwiające narządy trzewne (np. śledzionę i wątrobę) regulują ogólnoustrojową odpowiedź immunologiczną poprzez blokowanie aktywacji makrofagów i komórek NKT. W przypadku udaru i endotoksemii septycznej neurony te odgrywają rolę immunosupresyjną.
Zazwyczaj przebieg czasowy i charakter zapalenia, czy to podczas infekcji, reakcji alergicznych czy patologii autoimmunologicznych, jest definiowany przez kategorie zaangażowanych komórek odpornościowych. Ważne będzie, aby wiedzieć, jakie rodzaje komórek odpornościowych są regulowane przez sygnały czuciowe i autonomiczne. W odpowiedzi na to pytanie może pomóc systematyczna ocena tego, jakie mediatory mogą być uwalniane z nocyceptorów i neuronów autonomicznych oraz ekspresja receptorów dla nich przez różne wrodzone i nabyte komórki odpornościowe.
Podczas ewolucji podobne szlaki molekularne wykrywania zagrożeń rozwinęły się zarówno dla odporności wrodzonej, jak i nocycepcji, mimo że komórki mają zupełnie inne linie rozwojowe. Podczas gdy PRR i szkodliwe kanały jonowe bramkowane ligandami są badane oddzielnie przez immunologów i neurobiologów, granica między tymi dwoma obszarami jest coraz bardziej zacierana. Podczas uszkodzenia tkanek i infekcji patogennej, uwolnienie sygnałów zagrożenia prawdopodobnie doprowadzi do skoordynowanej aktywacji zarówno neuronów obwodowych, jak i komórek odpornościowych, ze złożoną komunikacją dwukierunkową i zintegrowaną obroną gospodarza. Anatomiczne umiejscowienie nocyceptorów na styku z otoczeniem, szybkość transdukcji neuronalnej i ich zdolność do uwalniania silnych koktajli mediatorów działających na układ odpornościowy pozwala obwodowemu układowi nerwowemu na aktywne modulowanie wrodzonej odpowiedzi immunologicznej i koordynację dalszej odporności nabytej. I odwrotnie, nocyceptory są bardzo wrażliwe na mediatory immunologiczne, które aktywują i uwrażliwiają neurony. Zapalenie neurogenne i immunologiczne nie są zatem niezależnymi jednostkami, ale działają razem jako urządzenia wczesnego ostrzegania. Jednak obwodowy układ nerwowy odgrywa również ważną rolę w patofizjologii i być może etiologii wielu chorób immunologicznych, takich jak astma, łuszczyca lub zapalenie jelita grubego, ponieważ jego zdolność do aktywacji układu odpornościowego może nasilać patologiczny stan zapalny 15�17. Leczenie zaburzeń immunologicznych może zatem wymagać ukierunkowania na nocyceptory, jak również na komórki odpornościowe.
Podziękowania
Dziękujemy NIH za wsparcie (2R37NS039518).
Podsumowując„Zrozumienie roli zapalenia neurogennego w obronie gospodarza i immunopatologii jest niezbędne do określenia właściwego podejścia do leczenia różnych problemów zdrowotnych układu nerwowego. Przyglądając się interakcjom neuronów obwodowych z komórkami odpornościowymi, pracownicy służby zdrowia mogą rozwijać podejścia terapeutyczne, aby jeszcze bardziej wzmocnić obronę gospodarza, a także zahamować immunopatologię. Celem powyższego artykułu jest pomoc pacjentom w zrozumieniu neurofizjologii klinicznej neuropatii, wśród innych problemów zdrowotnych związanych z uszkodzeniem nerwów. Informacje podane w National Center for Biotechnology Information (NCBI). Zakres naszych informacji ogranicza się do chiropraktyki oraz urazów i stanów kręgosłupa. Aby omówić ten temat, prosimy zapytać dr Jimeneza lub skontaktować się z nami pod adresem�915-850-0900 .
Kurator: dr Alex Jimenez
Dodatkowe tematy: Back Pain
Na ból pleców jest jedną z najczęstszych przyczyn niepełnosprawności i nieobecności w pracy na całym świecie. W rzeczywistości, ból pleców został przypisany jako drugi najczęstszy powód wizyt u lekarza, przewyższający jedynie infekcje górnych dróg oddechowych. Około 80 procent populacji doświadczy pewnego rodzaju bólu pleców przynajmniej raz w ciągu całego życia. Kręgosłup jest złożoną strukturą zbudowaną z kości, stawów, więzadeł i mięśni oraz innych miękkich tkanek. Z tego powodu urazy i / lub pogorszenie warunków, takich jak przepukliny, może ostatecznie prowadzić do objawów bólu pleców. Urazy sportowe lub wypadki samochodowe są często najczęstszą przyczyną bólu pleców, jednak czasami najprostsze ruchy mogą mieć bolesne skutki. Na szczęście alternatywne opcje leczenia, takie jak opieka chiropraktyczna, mogą pomóc złagodzić ból pleców poprzez zastosowanie korekcji kręgosłupa i manualnych manipulacji, ostatecznie poprawiając ulgę w bólu.
1.�Sauer SK, Reeh PW, Bove GM. Szkodliwe, indukowane ciepłem uwalnianie CGRP z aksonów nerwu kulszowego szczura in vitroEur J Neurosci.�2001;14:1203.�[PubMed]
2.�Edvinsson L, Ekman R, Jansen I, McCulloch J, Uddman R. Peptyd związany z genem kalcytoniny i mózgowe naczynia krwionośne: dystrybucja i efekty naczynioruchowe.J Cereb Metab przepływu krwi.�1987;7:720.�[PubMed]
3.�McCormack DG, Mak JC, Coupe MO, Barnes PJ. Związane z genem kalcytoniny rozszerzenie naczyń peptydowych ludzkich naczyń płucnychJ Appl Physiol.�1989;67:1265.�[PubMed]
4.�Saria A. Substancja P we włóknach nerwów czuciowych przyczynia się do rozwoju obrzęku tylnej łapy szczura po urazie termicznym.Br J. Pharmacol.�1984;82:217.�[Artykuł bezpłatny PMC][PubMed]
5.�Mózg SD, Williams TJ. Interakcje między tachykininami i peptydem genetycznym kalcytoniny prowadzą do modulacji powstawania obrzęków i przepływu krwi w skórze szczura.Br J. Pharmacol.�1989;97: 77 82.[Artykuł bezpłatny PMC][PubMed]
6.�Fryer AD i in. Eotaksyna neuronalna i wpływ antagonisty CCR3 na nadreaktywność dróg oddechowych i dysfunkcję receptora M2.J Clin Invest.�2006;116:228.�[Artykuł bezpłatny PMC][PubMed]
7.�Ansel JC, Brown JR, Payan DG, Brown MA. Substancja P selektywnie aktywuje ekspresję genu TNF-alfa w mysich komórkach tucznychJ Immunol.�1993;150:4478.�[PubMed]
8.�Ding W, Stohl LL, Wagner JA, Granstein RD. Peptyd związany z genem kalcytoniny skłania komórki Langerhansa do odporności typu Th2.J Immunol.�2008;181:6020.�[Artykuł bezpłatny PMC][PubMed]
9.�Hosoi J, et al. Regulacja funkcji komórek Langerhansa przez nerwy zawierające peptyd związany z genem kalcytoniny.�Natura.�1993;363:159.�[PubMed]
10.�Mikami N, et al. Peptyd związany z genem kalcytoniny jest ważnym regulatorem odporności skórnej: wpływ na funkcje komórek dendrytycznych i komórek T.J Immunol.�2011;186:6886.�[PubMed]
11.�Rochlitzer S, et al. Peptyd związany z genem kalcytoniny neuropeptydu wpływa na alergiczne zapalenie dróg oddechowych poprzez modulowanie funkcji komórek dendrytycznych.Clin Exp Alergia.�2011;41:1609.�[PubMed]
12.�Cyphert JM i in. Współpraca między komórkami tucznymi a neuronami jest niezbędna do skurczu oskrzeli, w którym pośredniczy antygen.�J Immunol.�2009;182:7430.�[Artykuł bezpłatny PMC][PubMed]
13.�Levine JD, et al. Substancja wewnątrzneuronowa P przyczynia się do nasilenia eksperymentalnego zapalenia stawów.�Nauka.�1984;226:547.�[PubMed]
14.�Levine JD, Khasar SG, Zielony PG. Zapalenie neurogenne i zapalenie stawów.�Ann NY Acad Sci.�2006;1069:155.�[PubMed]
15.�Engel MA i in. TRPA1 i substancja P pośredniczą w zapaleniu jelita grubego u myszy.�Gastroenterologia.�2011;141:1346.�[PubMed]
16.�Ostrowski SM, Belkadi A, Loyd CM, Diaconu D, Ward NL. Odnerwienie skórne łuszczycowej skóry myszy poprawia akantozę i stan zapalny w sposób zależny od neuropeptydów czuciowych.J Zainwestuj Dermatol.�2011;131:1530.�[Artykuł bezpłatny PMC][PubMed]
17.�Caceres AI, et al. Sensoryczny neuronalny kanał jonowy niezbędny do zapalenia dróg oddechowych i nadreaktywności w astmie.�Proc Natl Acad Sci USA A.�2009;106:9099.�[Artykuł bezpłatny PMC][PubMed]
18.�Caterina MJ i in. Upośledzenie nocycepcji i odczuwania bólu u myszy pozbawionych receptora kapsaicyny.�Nauka.�2000;288:306.�[PubMed]
19.�Bessac BF i in. Antagoniści ankyryny 1 o potencjale przejściowym blokują szkodliwe działanie toksycznych izocyjanianów przemysłowych i gazów łzawiących.FASEB J.�2009;23:1102.�[Artykuł bezpłatny PMC][PubMed]
20.�Cruz-Orengo L, et al. Nocycepcja skórna wywołana przez 15-delta PGJ2 poprzez aktywację kanału jonowego TRPA1.�Mol ból.�2008;4:30.�[Artykuł bezpłatny PMC][PubMed]
21.�Trevisani M i in. 4-hydroksynonenal, endogenny aldehyd, powoduje ból i neurogenne zapalenie poprzez aktywację receptora drażniącego TRPA1.�Proc Natl Acad Sci USA A.�2007;104:13519.�[Artykuł bezpłatny PMC][PubMed]
22.�Janeway CA, Jr, Medzhitov R. Wprowadzenie: rola odporności wrodzonej w adaptacyjnej odpowiedzi immunologicznej.Semin Immunol.�1998;10:349.�[PubMed]
23.�Matzinger P. Wrodzone poczucie zagrożenia.�Ann NY Acad Sci.�2002;961:341.�[PubMed]
24.�Bianchi MNIE. DAMP, PAMP i alarminy: wszystko, co musimy wiedzieć o niebezpieczeństwie.�J Leukoc Biol.�2007;81:1.�[PubMed]
25.�Liu T, Xu ZZ, Park CK, Berta T, Ji RR. Receptor Toll-podobny 7 pośredniczy w świądzie.�Nat Neurosci.�2010;13:1460.�[Artykuł bezpłatny PMC][PubMed]
26.�Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. LPS uwrażliwia TRPV1 poprzez aktywację TLR4 w trójdzielnych neuronach czuciowych.�J Dent Res.�2011;90:759.�[PubMed]
27.�Qi J, et al. Bolesne ścieżki indukowane przez stymulację TLR neuronów zwoju korzenia grzbietowego.�J Immunol.�2011;186:6417.�[Artykuł bezpłatny PMC][PubMed]
28.�Cockayne DA i in. Hiprefleksja pęcherza moczowego i zmniejszone zachowanie związane z bólem u myszy z niedoborem P2X3.�Natura.�2000;407:1011.�[PubMed]
29.�Mariathasan S, et al. Kriopiryna aktywuje inflamasom w odpowiedzi na toksyny i ATP.�Natura.�2006;440:228.�[PubMed]
30.�Souslova V, et al. Deficyty kodowania ciepła i nieprawidłowy ból zapalny u myszy pozbawionych receptorów P2X3.�Natura.�2000;407:1015.�[PubMed]
31.�de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. Platforma molekularna w neuronach reguluje stan zapalny po urazie rdzenia kręgowego.�J. Neurosci.�2008;28:3404.�[PubMed]
32.�LinkTM, et al. TRPV2 odgrywa kluczową rolę w wiązaniu cząstek makrofagów i fagocytozy.�Nat Immunol.�2010;11:232.�[Artykuł bezpłatny PMC][PubMed]
33.�Turner H, del Carmen KA, Stokes A. Połączenie między kanałami TRPV a funkcją mast cell.�Handb Exp Pharmacol.�2007: 457.�[PubMed]
34.�Binshtok AM i in. Nocyceptory to czujniki interleukiny-1beta.�J. Neurosci.�2008;28: 14062 14073.[Artykuł bezpłatny PMC][PubMed]
35.�Zhang XC, Kainz V, Burstein R, Levy D. Czynnik martwicy nowotworu-alfa indukuje uczulenie nocyceptorów opon mózgowych za pośrednictwem lokalnych działań kinazy COX i p38 MAP.Ból.�2011;152: 140 149.[Artykuł bezpłatny PMC][PubMed]
36.�Samad TA i in. Zależna od interleukiny-1beta indukcja Cox-2 w OUN przyczynia się do nadwrażliwości na ból zapalny.Natura.�2001;410:471.�[PubMed]
37.�Veres TZ i in. Przestrzenne interakcje między komórkami dendrytycznymi a nerwami czuciowymi w alergicznym zapaleniu dróg oddechowych.�Am J Respir Cell Mol Biol2007;37:553.�[PubMed]
38.�Smith CH, Barker JN, Morris RW, MacDonald DM, Lee TH. Neuropeptydy indukują szybką ekspresję cząsteczek adhezyjnych komórek śródbłonka i wywołują naciek granulocytów w ludzkiej skórze.J Immunol.�1993;151:3274.�[PubMed]
40.�Ganor Y, Besser M, Ben-Zakay N, Unger T, Levite M. Ludzkie komórki T wyrażają funkcjonalny jonotropowy receptor glutaminianu GluR3, a sam glutaminian wyzwala adhezję za pośrednictwem integryny do lamininy i fibronektyny oraz migrację chemotaktyczną.J Immunol.�2003;170:4362.�[PubMed]
41.�Czepielewski RS, et al. Receptor peptydowy uwalniający gastrynę (GRPR) pośredniczy w chemotaksji w neutrofilach.Proc Natl Acad Sci USA A.�2011;109:547.�[Artykuł bezpłatny PMC][PubMed]
42.�Brogden KA, Guthmiller JM, Salzet M, Zasloff M. Układ nerwowy i odporność wrodzona: połączenie neuropeptydów.Nat Immunol.�2005;6:558.�[PubMed]
43.�Jimeno R i in. Wpływ VIP na równowagę między cytokinami a głównymi regulatorami aktywowanych limfocytów T pomocniczych.�Immunol Komórka Biol.�2011;90:178.�[PubMed]
44.�Razavi R i in. Neurony czuciowe TRPV1+ kontrolują stres komórek beta i zapalenie wysp w cukrzycy autoimmunologicznejKomórka.�2006;127:1123.�[PubMed]
45.�Cunin P i in. Tachykininy, substancja P i hemokinina-1, sprzyjają wytwarzaniu ludzkich komórek pamięci Th17 poprzez indukcję ekspresji IL-1beta, IL-23 i TNF-podobnego 1A przez monocyty.J Immunol.�2011;186:4175.�[PubMed]
46.�Andersson U, Tracey KJ. Odruchowe zasady homeostazy immunologicznej.�Annu Rev Immunol.�2011[Artykuł bezpłatny PMC][PubMed]
47.�de Jonge WJ, et al. Stymulacja nerwu błędnego osłabia aktywację makrofagów poprzez aktywację ścieżki sygnałowej Jak2-STAT3.Nat Immunol.�2005;6:844.�[PubMed]
48.�Rosas-Ballina M, et al. Komórki T syntetyzujące acetylocholinę przekazują sygnały nerwowe w obwodzie nerwu błędnegoNauka.�2011;334:98.�[Artykuł bezpłatny PMC][PubMed]
49.�Wang H i in. Podjednostka alfa7 nikotynowego receptora acetylocholiny jest niezbędnym regulatorem stanu zapalnego.�Natura.�2003;421:384.�[PubMed]
50.�Wong CH, Jenne CN, Lee WY, Leger C, Kubes P. Funkcjonalne unerwienie komórek iNKT wątroby działa immunosupresyjnie po udarze.Nauka.�2011;334:101.�[PubMed]
Narzędzie Find A Practitioner firmy IFM to największa sieć skierowań w medycynie funkcjonalnej, stworzona, aby pomóc pacjentom zlokalizować lekarzy medycyny funkcjonalnej na całym świecie. Certyfikowani praktycy IFM są wymienieni na pierwszym miejscu w wynikach wyszukiwania ze względu na ich rozległe wykształcenie w zakresie medycyny funkcjonalnej



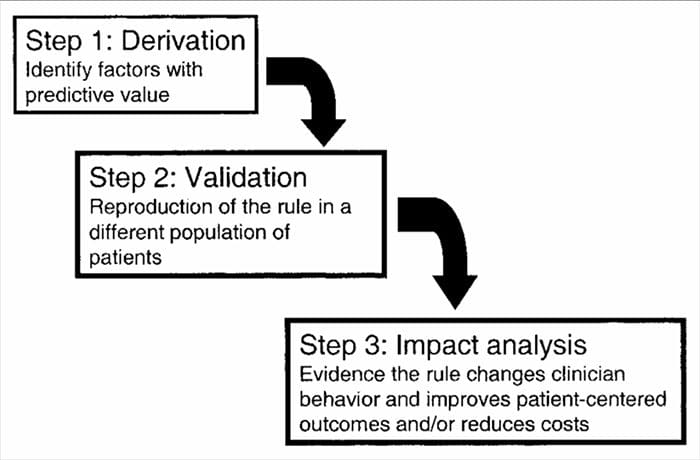




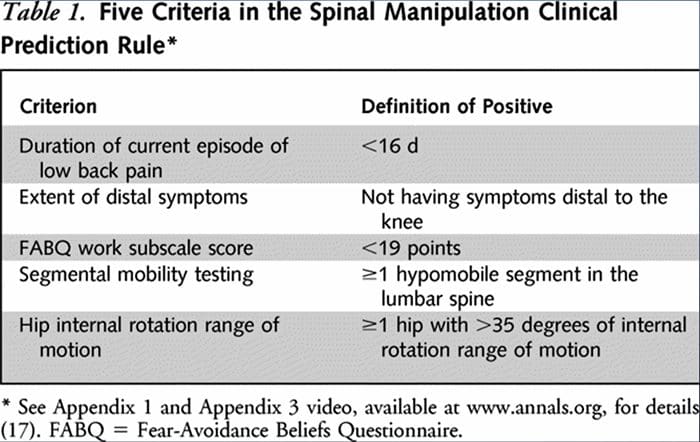
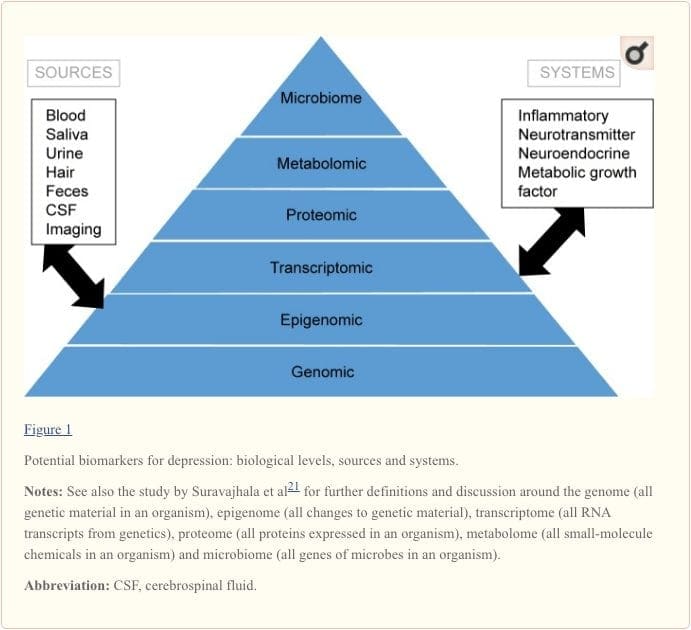





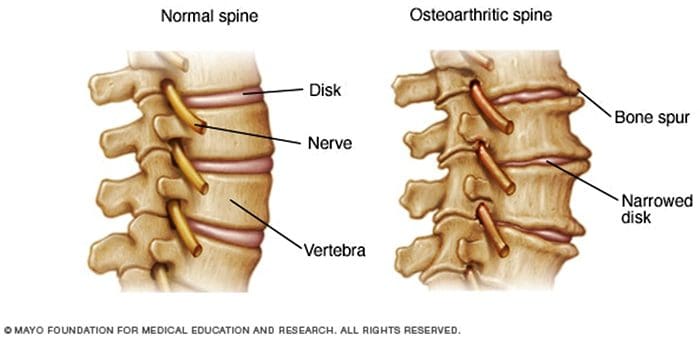
 Abstrakcyjny
Abstrakcyjny
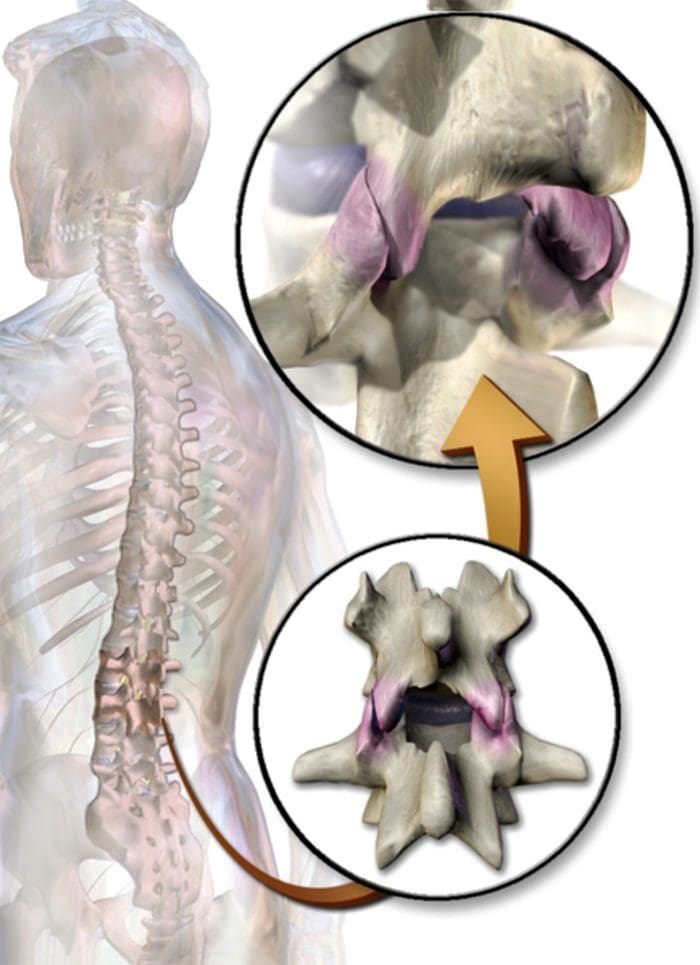
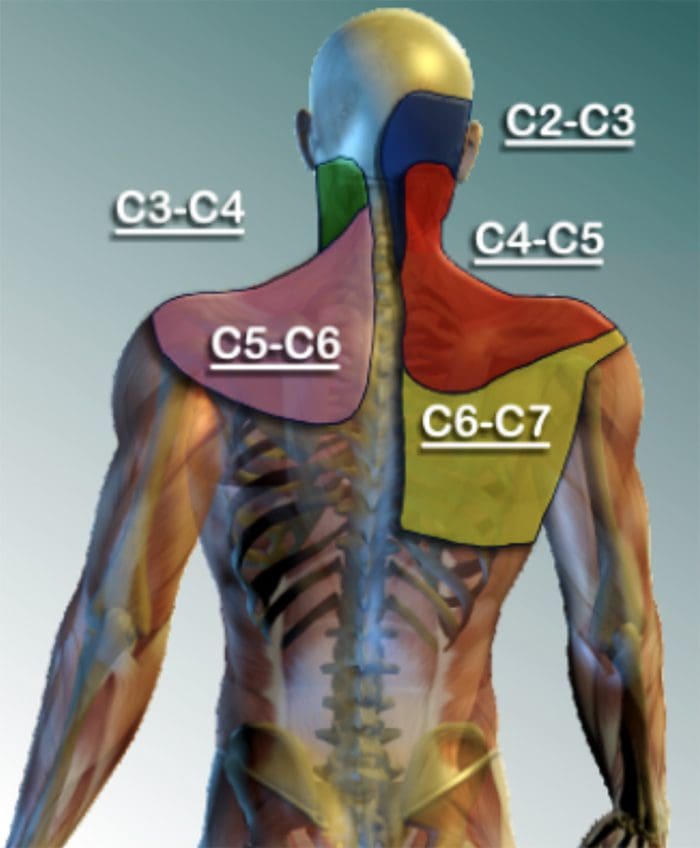
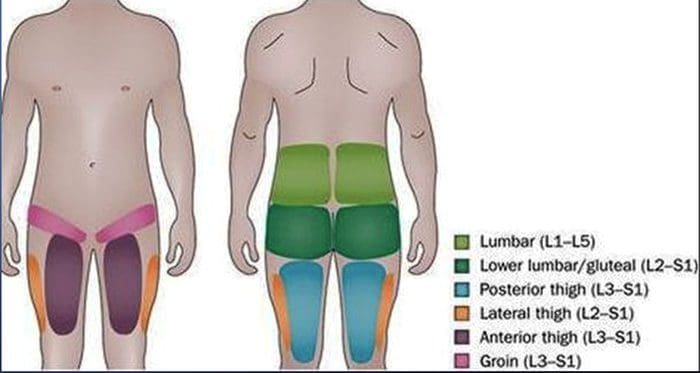

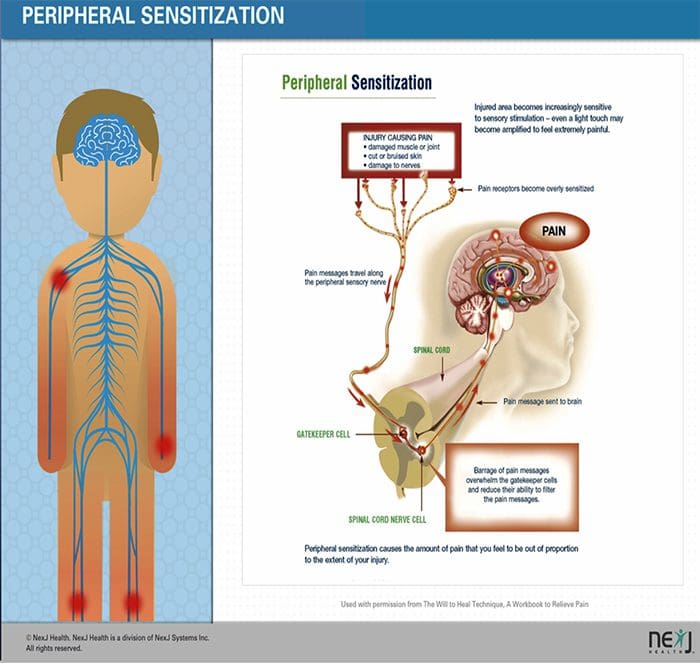
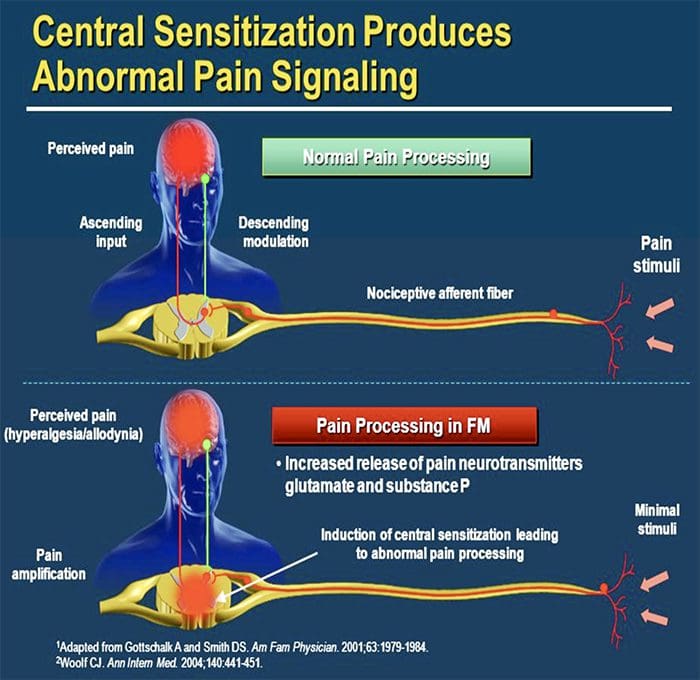
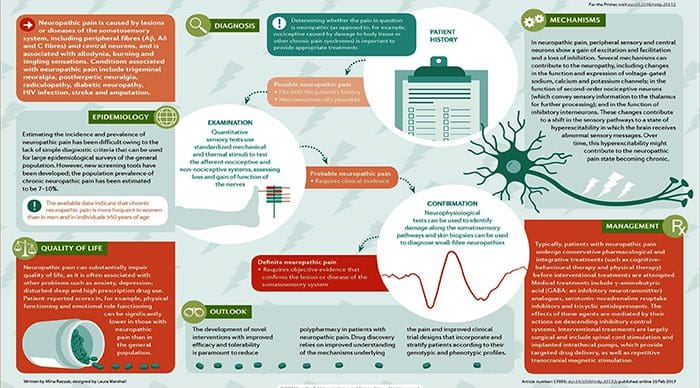


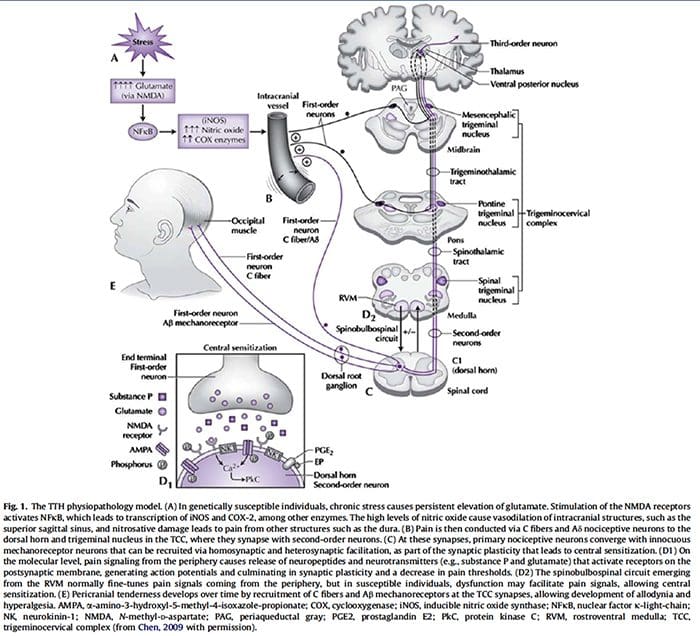

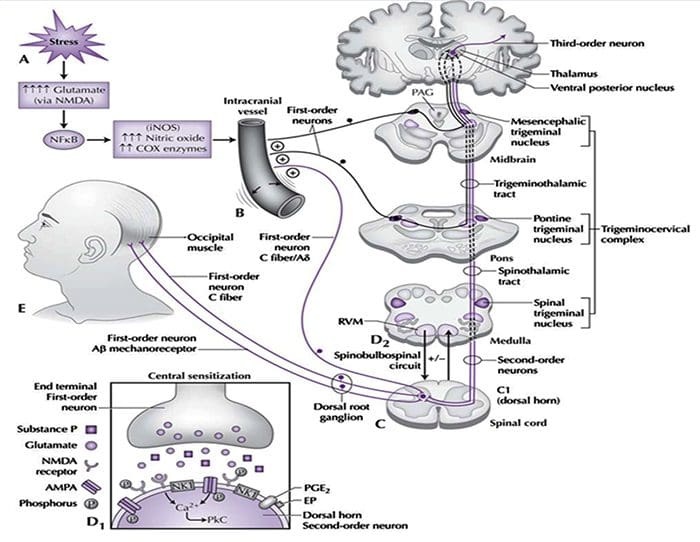












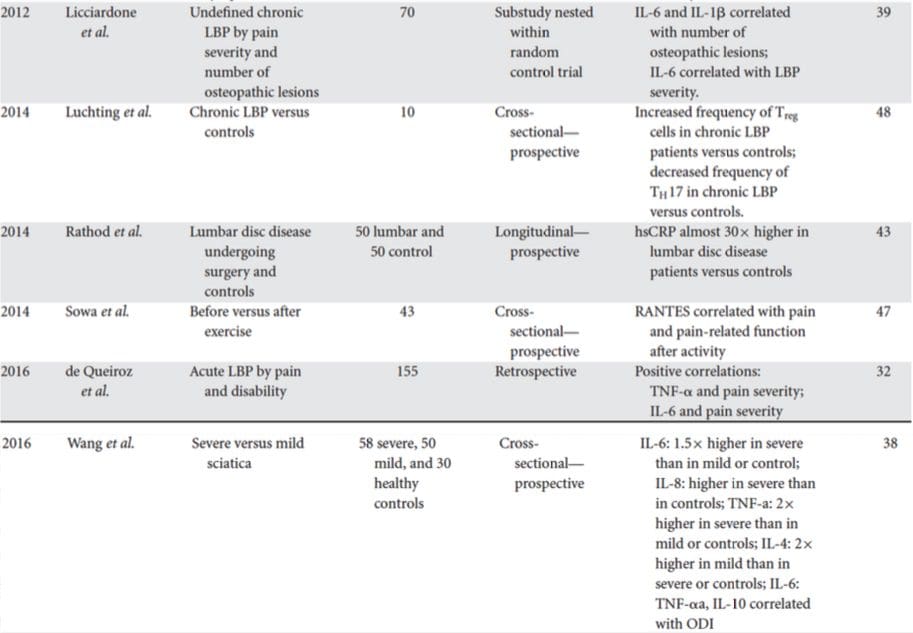


 Referencje:
Referencje:
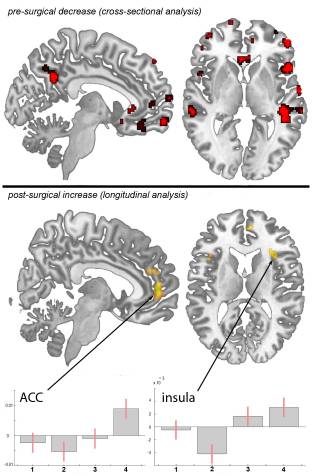
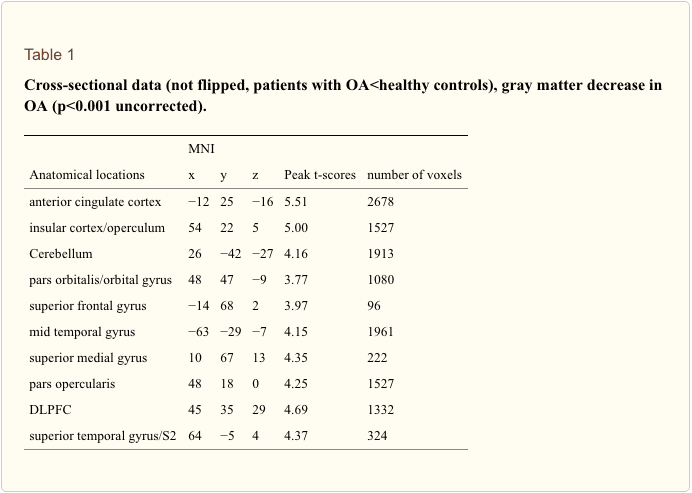
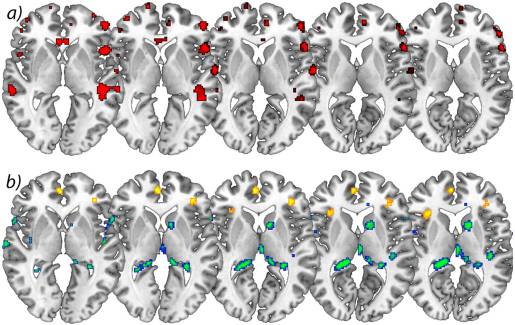
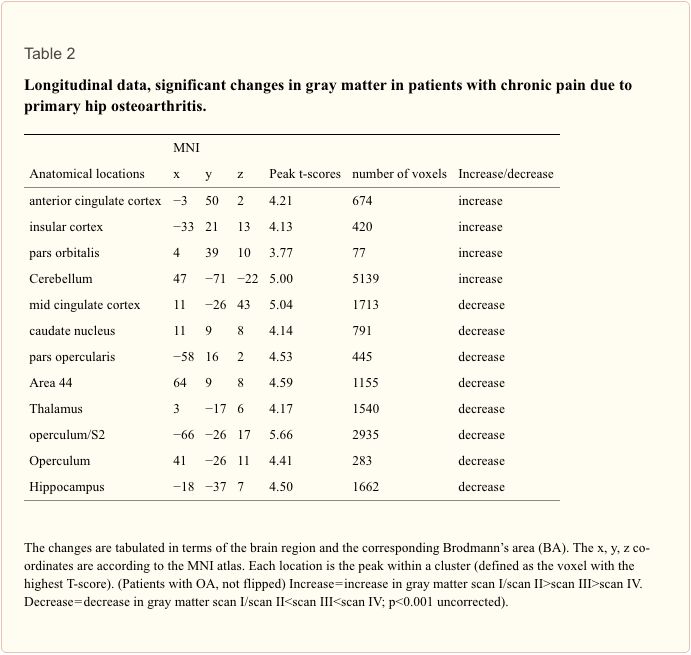
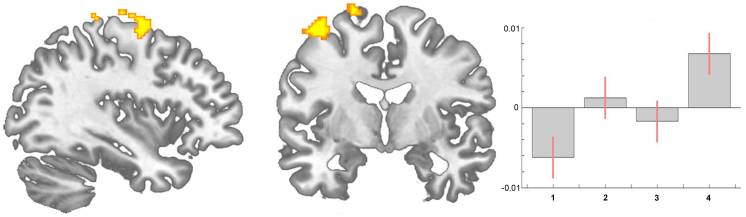
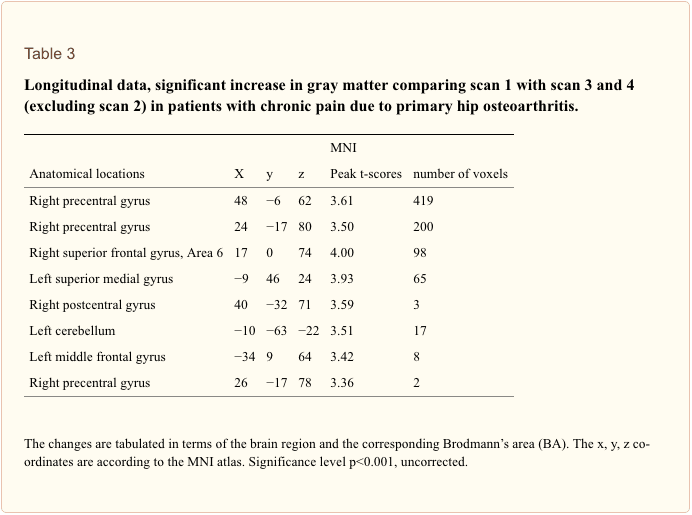

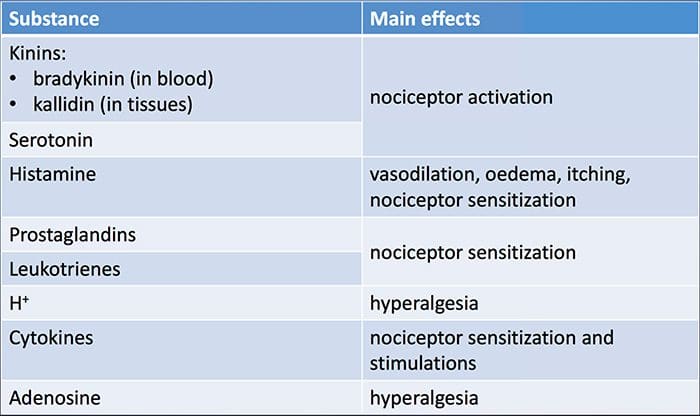
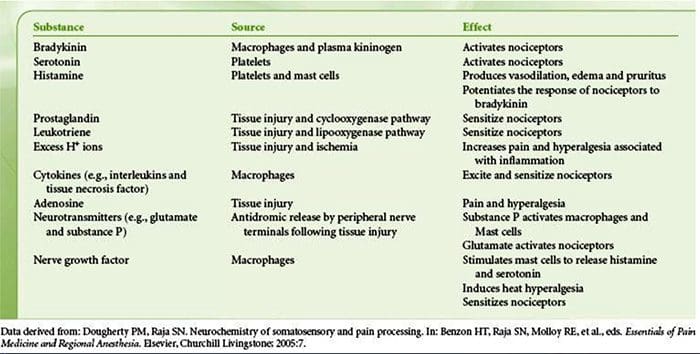
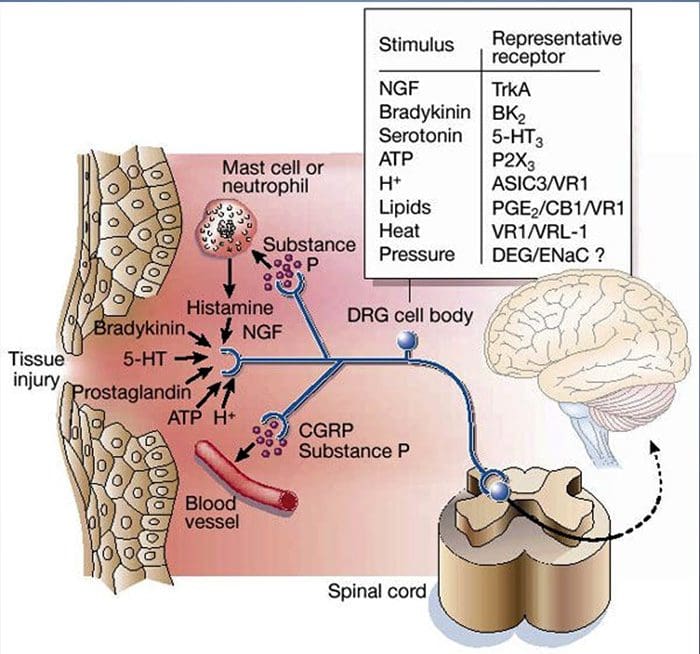
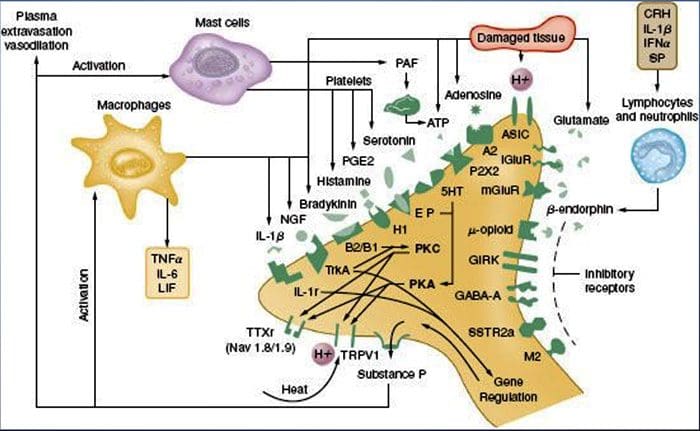 Dlaczego boli mnie ramię? Przegląd neuroanatomicznej i biochemicznej podstawy bólu barku
Dlaczego boli mnie ramię? Przegląd neuroanatomicznej i biochemicznej podstawy bólu barku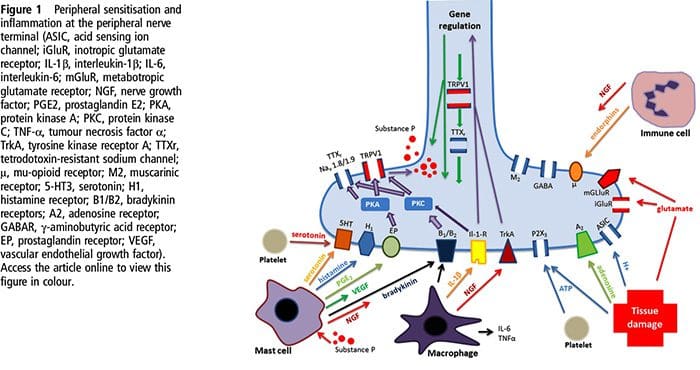 NGF i receptor członu 1 kanału kationowego podrodziny V podrodziny receptora przejściowego potencjalnego receptora (TRPV1) mają symbiotyczny związek, jeśli chodzi o zapalenie i uczulenie na receptory nocyceptorowe. Cytokiny wytwarzane w tkance w stanie zapalnym powodują wzrost produkcji NGF.19 NGF stymuluje uwalnianie histaminy i serotoniny (5-HT3) przez komórki tuczne, a także uwrażliwia nocyceptory, prawdopodobnie zmieniając właściwości A? włókna tak, że większa część staje się nocyceptywna. Receptor TRPV1 jest obecny w subpopulacji pierwotnych włókien doprowadzających i jest aktywowany przez kapsaicynę, ciepło i protony. Receptor TRPV1 jest syntetyzowany w ciele komórki włókna aferentnego i jest transportowany zarówno do terminali obwodowych, jak i centralnych, gdzie przyczynia się do wrażliwości neuronów aferentnych nocyceptywnych. Zapalenie powoduje obwodowe wytwarzanie NGF, który następnie wiąże się z receptorem kinazy tyrozynowej typu 1 na końcach nocyceptorów, NGF jest następnie transportowany do ciała komórki, gdzie prowadzi do regulacji w górę transkrypcji TRPV1, a w konsekwencji do zwiększonej wrażliwości na receptory nocyceptorowe.19 20 NGF i inne mediatory zapalne również uwrażliwiają TRPV1 poprzez różnorodną gamę drugorzędowych szlaków przekaźnikowych. Uważa się, że wiele innych receptorów, w tym receptory cholinergiczne, receptory kwasu y-aminomasłowego (GABA) i receptory somatostatyny, są również zaangażowane w wrażliwość obwodowych receptorów nocyceptorowych.
NGF i receptor członu 1 kanału kationowego podrodziny V podrodziny receptora przejściowego potencjalnego receptora (TRPV1) mają symbiotyczny związek, jeśli chodzi o zapalenie i uczulenie na receptory nocyceptorowe. Cytokiny wytwarzane w tkance w stanie zapalnym powodują wzrost produkcji NGF.19 NGF stymuluje uwalnianie histaminy i serotoniny (5-HT3) przez komórki tuczne, a także uwrażliwia nocyceptory, prawdopodobnie zmieniając właściwości A? włókna tak, że większa część staje się nocyceptywna. Receptor TRPV1 jest obecny w subpopulacji pierwotnych włókien doprowadzających i jest aktywowany przez kapsaicynę, ciepło i protony. Receptor TRPV1 jest syntetyzowany w ciele komórki włókna aferentnego i jest transportowany zarówno do terminali obwodowych, jak i centralnych, gdzie przyczynia się do wrażliwości neuronów aferentnych nocyceptywnych. Zapalenie powoduje obwodowe wytwarzanie NGF, który następnie wiąże się z receptorem kinazy tyrozynowej typu 1 na końcach nocyceptorów, NGF jest następnie transportowany do ciała komórki, gdzie prowadzi do regulacji w górę transkrypcji TRPV1, a w konsekwencji do zwiększonej wrażliwości na receptory nocyceptorowe.19 20 NGF i inne mediatory zapalne również uwrażliwiają TRPV1 poprzez różnorodną gamę drugorzędowych szlaków przekaźnikowych. Uważa się, że wiele innych receptorów, w tym receptory cholinergiczne, receptory kwasu y-aminomasłowego (GABA) i receptory somatostatyny, są również zaangażowane w wrażliwość obwodowych receptorów nocyceptorowych.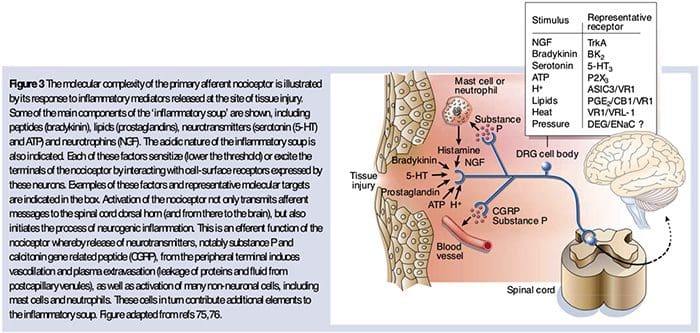 Neurochemia nocyceptorów
Neurochemia nocyceptorów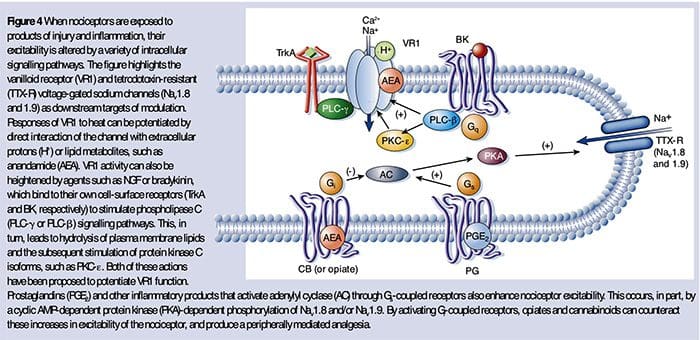 Komórkowe i molekularne mechanizmy bólu
Komórkowe i molekularne mechanizmy bólu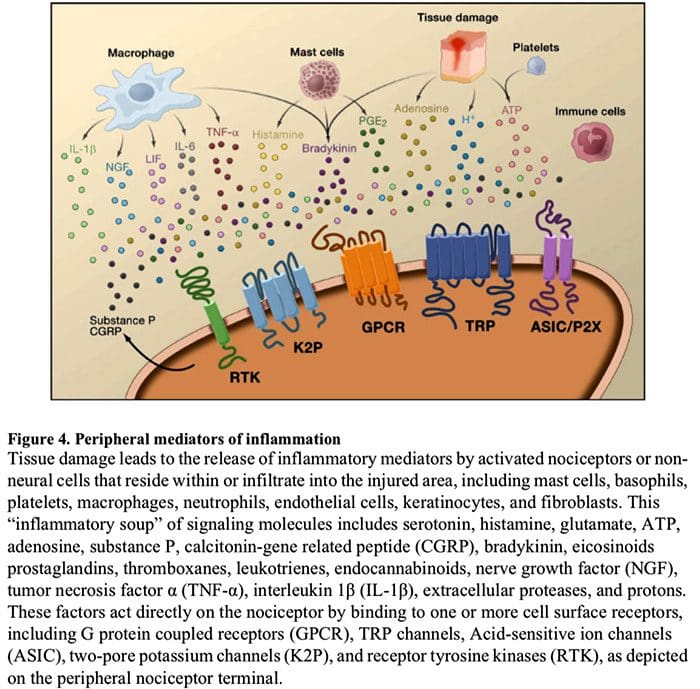
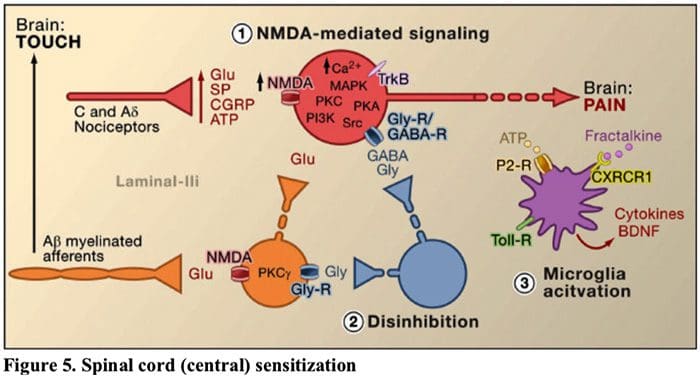 Rycina 5. Uczulenie rdzenia kręgowego (ośrodkowe)
Rycina 5. Uczulenie rdzenia kręgowego (ośrodkowe)