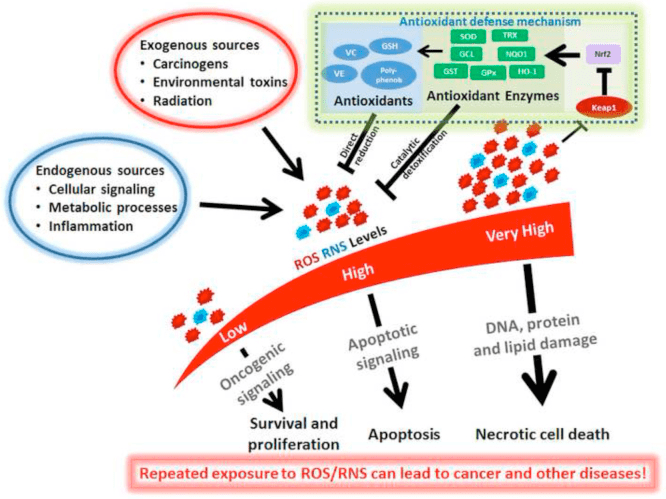
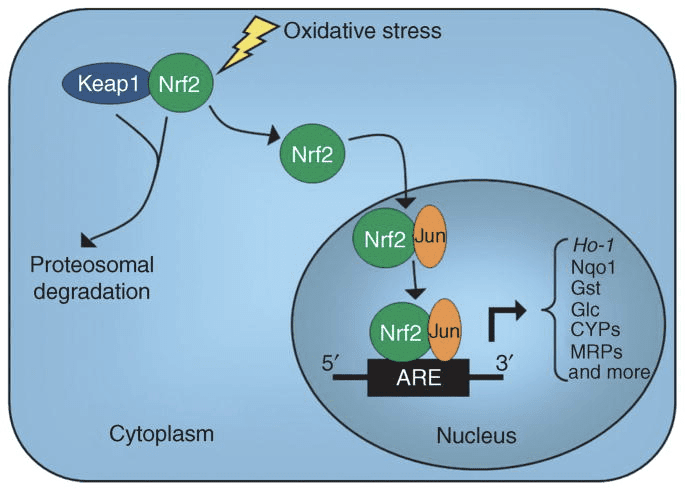
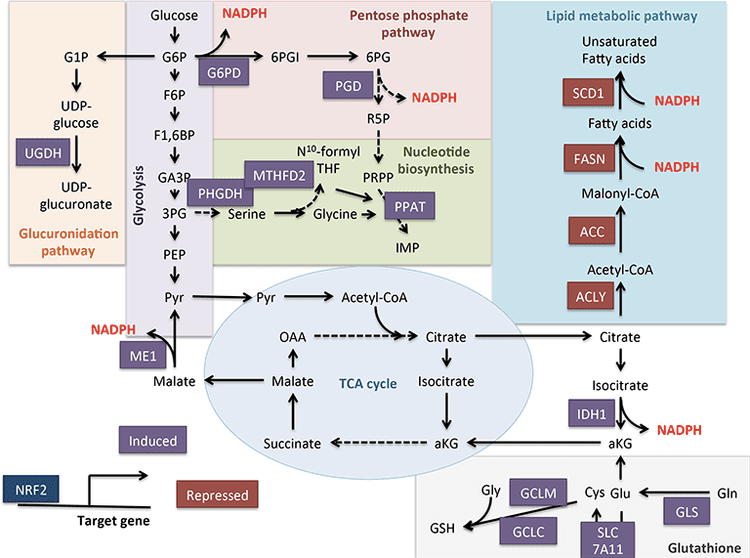
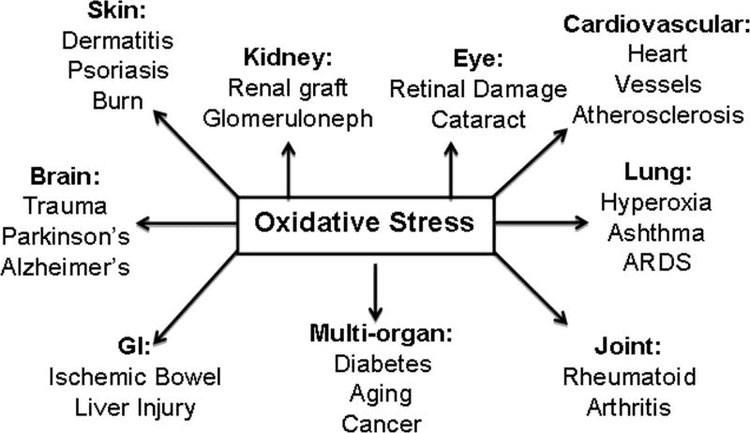
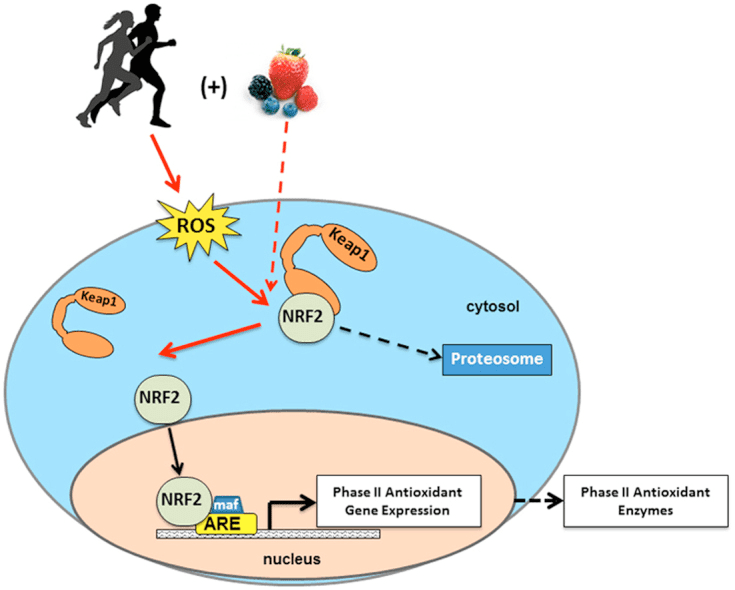
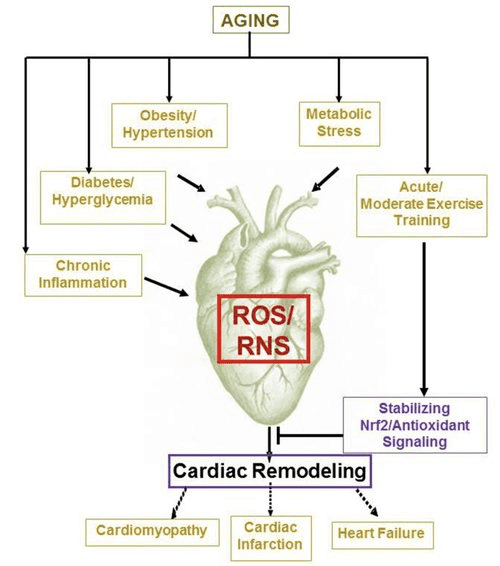
Powrót Klinika Chiropraktyka i Zespół Medycyny Funkcjonalnej Stresu Oksydacyjnego. Stres oksydacyjny definiuje się jako zaburzenie równowagi między produkcją reaktywnego tlenu (wolnych rodników) a obroną antyoksydacyjną. Innymi słowy, jest to brak równowagi między produkcją wolnych rodników a zdolnością organizmu do przeciwdziałania lub detoksykacji szkodliwych skutków poprzez neutralizację przez antyoksydanty. Stres oksydacyjny prowadzi do wielu stanów patofizjologicznych w organizmie. Obejmują one choroby neurodegeneracyjne, tj. chorobę Parkinsona, chorobę Alzheimera, mutacje genów, nowotwory, zespół przewlekłego zmęczenia, zespół łamliwego chromosomu X, zaburzenia serca i naczyń krwionośnych, miażdżycę, niewydolność serca, zawał serca i choroby zapalne. Utlenianie ma miejsce w wielu okolicznościach:
komórki wykorzystują glukozę do wytwarzania energii
układ odpornościowy zwalcza bakterie i tworzy stan zapalny
organizmy odtruwają zanieczyszczenia, pestycydy i dym papierosowy
W naszych ciałach zachodzą miliony procesów, które mogą prowadzić do utleniania. Oto kilka symptomów:
Zmęczenie
Utrata pamięci i/lub mgła mózgowa
Ból mięśni i/lub stawów
Zmarszczki wraz z siwymi włosami
Pogorszenie wzroku
Bóle głowy i wrażliwość na hałas
Podatność na infekcje
Wybór żywności ekologicznej i unikanie toksyn w środowisku ma duże znaczenie. To, wraz ze zmniejszeniem stresu, może być korzystne w zmniejszaniu utleniania.
Budzisz się zmęczony nawet po sześciu lub więcej godzinach snu?
Pod dużym stresem?
Jeśli doświadczasz którejkolwiek z tych sytuacji, może to być spowodowane poziomem melatoniny i kortyzolu wpływającym na twoje ciało i rytm dobowy.
Na całym świecie miliony ludzi mają problemy ze snem. W Stanach Zjednoczonych jest około 50-70 milionów ludzi którzy mają słabą jakość snu. Kiedy dana osoba spała krócej niż osiem godzin, staje się zmęczona i może pojawić się wiele problemów, zwłaszcza jeśli ich życie jest gorączkowe. Przy gorączkowym trybie życia i złym śnie może to spowodować, że organizm będzie miał mało energii, aby wykonać każde zadanie, poziom hormonu stresu kortyzolu zostanie podwyższony, a choroby takie jak wysokie ciśnienie krwi i cukrzyca mogą powodować problemy, które mogą być przewlekłe, jeśli nie są leczony.
W endokrynologii czynnościowej melatonina i kortyzol są hormonami wytwarzanymi przez organizm w sposób naturalny. Hormon kortyzolu lub hormon stresu pomaga organizmowi przejść w tryb „walki lub ucieczki”, co może być dobrą rzeczą dla każdego, kto wykonuje projekt lub idzie na rozmowę o pracę. Chociaż wysoki poziom hormonu kortyzolu może prowadzić do powikłań, takich jak stany zapalne, przewlekły stres oksydacyjny i wysokie ciśnienie krwi.
Dobowy rytm melatoniny
Dzięki hormonowi melatoniny, ten hormon informuje organizm, kiedy nadszedł czas na sen. Czasami jednak ludzie mają trudności ze snem, a przyjmowanie suplementów melatoniny może w rzeczywistości rozluźnić ciało, a tym samym sprawić, że osoba zasnie. Ponieważ szyszynka wytwarza melatoninę z mózgu, można ją również znaleźć w oczach, szpiku kostnym i jelitach, aby rozluźnić ciało i spowodować naturalny sen. Niektóre Badania pokazują że rytm dobowy szyszynki produkującej melatoninę. W ten sposób badania pokazują, że podawanie melatoniny może:
jeden: wywoływać sen u osób, które mają problemy z zasypianiem.
dwa: hamuje naturalne wybudzanie organizmu ze stymulatora rytmu dobowego.
Trzy: przesuń dobowy zegar biologiczny, aby zwiększyć spożycie snu, gdy osoba próbuje zasnąć o wcześniejszej porze, aby uzyskać pełne ośmiogodzinne korzyści ze snu.
Kiedy dana osoba pracuje w pracy od 9 do 5, podnosi się z ciałem i relaksuje ciało po ciężkim dniu pracy. Badania się dowiedziały że hormony melatoniny i kortyzolu pomagają regulować 24-godzinny wzorzec funkcjonowania organizmu i jego odpowiedzi. Cykl produkcji hormonów w organizmie może być zakłócony, jeśli dana osoba nie śpi późno w nocy lub śpi w ciągu dnia. Kiedy tak się dzieje, osoba może mieć destrukcyjne zaburzenia, takie jak wahania nastroju, zawroty głowy, być drażliwa i przygnębiona oraz mieć zaburzenia metaboliczne. Nie tylko to, ale również układ odpornościowy organizmu i jego układ hormonalny może również zostać uszkodzony, powodując, że organizm jest gospodarzem infekcji i chorób.
Przeprowadzono więcej badań dotyczących rytmów dobowych w ciele, ponieważ badania pokazują jak ludzie pracujący na nocnej zmianie są kojarzeni z ogromną liczbą niekorzystnych problemów zdrowotnych, które atakują układ sercowo-naczyniowy i pokarmowy, a także zaburzają układ metaboliczny. Każdy, kto pracował na nocną zmianę, musi zmienić swój harmonogram snu i dostosować się do szybkiej reorientacji harmonogramu snu/budzenia, aby iść do pracy i wykonywać swoją pracę. Ponieważ każdy pracuje w systemie zmianowym, może to być stresujące i może wpływać na wydajność organizmu pracownika, a także wpływać na wydzielanie melatoniny i kortyzolu.
Sposoby na wsparcie kortyzolu i melatoniny
Co zaskakujące, istnieją sposoby na obniżenie poziomu kortyzolu i upewnienie się, że poziom melatoniny działa prawidłowo, aby organizm mógł funkcjonować. Aby obniżyć poziom kortyzolu, należy wykonywać praktyki medytacyjne, znaleźć przyjemne hobby, a co najważniejsze, spróbować ćwiczeń głębokiego oddychania, aby rozluźnić ciało przed niechcianym stresem. Dzięki ćwiczeniom głębokiego oddychania może pomóc ciału uwolnić wszelkie napięcie, które utrzymuje dana osoba, a mięśnie ciała zaczynają się rozluźniać, a krew zaczyna płynąć. Dzięki poziomowi melatoniny współpracują one z rytmem dobowym organizmu i zapewniają, że organizm wie, kiedy nadszedł czas, aby się obudzić, spać i jeść. Hormon melatoniny może również pomóc w regulacji temperatury ciała, ciśnienie krwii poziomy hormonów, aby upewnić się, że działa prawidłowo. Gdy występuje wysoki poziom tych systemów, może to spowodować rozwój chorób przewlekłych i tym samym zaszkodzić ciału.
Badania pokazują że hormony melatoniny mogą wiązać się z receptorami neurologicznymi w organizmie, promując w ten sposób relaksację. Ponieważ melatonina wiąże się z receptorami neurologicznymi, może również zmniejszać aktywność nerwów i poziom dopaminy, powodując ciężkość oczu, przez co osoba zasypia.
Wnioski
Ponieważ organizm jest w stanie naturalnie wytwarzać poziomy melatoniny i kortyzolu, aby upewnić się, że organizm nie jest nadmiernie zestresowany przez cały dzień. Ponieważ melatonina współgra z rytmem dobowym organizmu, organizm wie, kiedy nie spać i zasnąć. Ponieważ każdy ma napięty harmonogram, ważne jest, aby poświęcić czas i zrelaksować się oraz zadbać o zdrowy harmonogram snu, aby ciało było zdrowe i funkcjonowało. Niektóre CYFROWE DLA MILIONÓW są tutaj, aby upewnić się, że układ hormonalny działa prawidłowo i wspiera nadnercza oraz metabolizm cukrów.
Zakres naszych informacji ogranicza się do zagadnień związanych z chiropraktyką, zdrowiem mięśniowo-szkieletowym i nerwowym oraz artykułów, tematów i dyskusji na temat medycyny funkcjonalnej. Używamy funkcjonalnych protokołów zdrowotnych w leczeniu urazów lub zaburzeń układu mięśniowo-szkieletowego. Nasze biuro podjęło uzasadnioną próbę dostarczenia wspierających cytatów i zidentyfikowało odpowiednie badanie lub badania potwierdzające nasze posty. Na żądanie udostępniamy również kopie badań pomocniczych. Aby dalej omawiać powyższy temat, możesz poprosić dr Alexa Jimeneza lub skontaktować się z nami pod adresem 915-850-0900.
Referencje:
Cajochen, C, i in. „Rola melatoniny w regulacji rytmów dobowych i snu człowieka”. Journal of Neuroendocrinology, US National Library of Medicine, kwiecień 2003, www.ncbi.nlm.nih.gov/pubmed/12622846.
James, Francine O, et al. „Dobowe rytmy ekspresji melatoniny, kortyzolu i zegarowej ekspresji genów podczas symulowanej pracy na nocne zmiany”. senAssociated Professional Sleep Societies, LLC, listopad 2007, www.ncbi.nlm.nih.gov/pmc/articles/PMC2082093/.
Monteleone, P, et al. �Związek czasowy między reakcjami melatoniny i kortyzolu na nocny stres fizyczny u ludzi.� Psychoneuroendocrinology, US National Library of Medicine, 1992, www.ncbi.nlm.nih.gov/pubmed/1609019.
Ramana, Ryana. �Jak melatonina może pomóc Ci spać i czuć się lepiej� HEALTHLINE, Healthline Media, 3 września 2017 r., www.healthline.com/nutrition/melatonin-and-sleep.
Zamanian, Zahra i in. „Zarys zmian rytmu dobowego kortyzolu i melatoniny u pracowników ochrony Uniwersytetu Medycznego w Shiraz”. International Journal of Preventive Medicine, Medknow Publications & Media Pvt Ltd, lipiec 2013, www.ncbi.nlm.nih.gov/pmc/articles/PMC3775223/.
Nowoczesna medycyna integracyjna i funkcjonalna Esse Quam Videri
Informując osoby o tym, w jaki sposób Narodowy Uniwersytet Nauk o Zdrowiu zapewnia wiedzę przyszłym pokoleniom, które chcą coś zmienić na świecie. Uniwersytet oferuje szeroki zakres zawodów medycznych dla medycyny funkcjonalnej i integracyjnej.
Czy zastanawiałeś się kiedyś, dlaczego czujesz się ospały po długim dniu? A może czujesz mdłości, gdy zjadłeś coś złego lub przesadziłeś z ulubionym jedzeniem? Czy to możliwe, że Twoje jelita wykazują oznaki stresu i dyskomfortu z powodu pewnych nawyków, z którymi możesz się spotkać, o których nawet nie wiedziałeś?
W naszym poprzednim artykule rozmawialiśmy sześć rodzajów żywności że nasze jelita muszą być zdrowe. Od naszego jelit zawiera biliony mikrobiomów, zarówno dobrych, jak i złych, mikrobiomy te odgrywają ważną rolę w naszym ogólnym zdrowiu. Zdrowy mikrobiom poprawia nasz zdrowie jelit, zdrowie serca, zdrowie mózgu, kontroluje naszą wagę i reguluje poziom cukru we krwi. Dzięki dobrym bakteriom w jelitach bakterie zapewniają nam dobry układ trawienny i niszczą szkodliwe bakterie. Ale niektóre style życia i wybory żywieniowe mogą faktycznie zwiększyć liczbę złych bakterii i obniżyć dobre bakterie i ogólny stan zdrowia.
Oto pięć zaskakująco wyborów stylu życia, które szkodzą jelitowi:
Nie jeść szerokiej gamy produktów spożywczych
Nasze jelita odgrywają ważną rolę w naszym ogólnym zdrowiu. Kiedy jemy dobre całe jedzenie, nasze jelita są szczęśliwsze; mamy więcej energii, aby wykonać każde zadanie, które zostanie nam powierzone i otrzymujemy składniki odżywcze dla naszej flory jelitowej. Jednak w ciągu ostatnich kilku dziesięcioleci bardziej skupialiśmy się na żywności przetworzonej ze względu na presję ekonomiczną związaną ze zwiększoną produkcją żywności. FOA stwierdził, że 75% żywności na świecie pochodzi tylko z 12 roślin i pięciu gatunków zwierząt, co jest bardzo szkodliwe dla flory jelitowej.
W klinice Injury Medical & Chiropractic Clinic informujemy naszych pacjentów o tym, jak ważne jest spożywanie pełnowartościowych, pełnowartościowych pokarmów, aby promować nie tylko zdrowe jelita, ale także zdrowy umysł. Kiedy ciało zostanie wprowadzone do pliku szeroka gama całych produktów spożywczych (z wysoką zawartością błonnika), nasze jelita zaczynają naprawiać uszkodzenia przetworzonej żywności, które mogliśmy spożyć wewnętrznie.
Jeśli jednak lekceważysz prebiotyki w swojej diecie, to jesteś szkodząc zdrowiu trawiennemu. Bez prebiotyków nasz układ trawienny spowalnia rozwój i różnorodność flory jelitowej. Więc aby mieć zdrowy mikrobiom rozwoju, musisz włączyć do swojej diety pokarmy wypełnione zarówno strawnymi, jak i niestrawnymi włóknami. Niektóre produkty spożywcze zawarte w tej kategorii to owies, orzechy, cebula, czosnek, pory, szparagi, banany, gruszki, ciecierzyca i fasola.
Trzymanie się diety bogatej w błonnik może być trudne, jednak istnieje możliwość przyjmowania suplementów prebiotycznych. Jeśli masz alergen pokarmowy lub wrażliwość pokarmową na pokarmy bogate w błonnik, weź suplementy prebiotyczne może faktycznie pomóc w rozwoju Bifidobacterium i Faecalibacterium w jelitach i być korzystny dla zdrowia bez dyskomfortu.
Nadmierne spożycie alkoholu
Każdy dorosły od czasu do czasu pije alkohol. Tak, to jeden z tych napojów, który pomaga trochę się zrelaksować po długim dniu, jednak zbyt duża jego ilość może prowadzić do nadużywania i uzależnienia od alkoholu. Czy wiesz, że spożywanie takiej ilości alkoholu jest szkodliwe twoje serce, wątroba i mózg; w ten sposób szkodzi zdrowiu jelit i powoduje dysbiozę?
Jedno badanie stwierdzili, że alkoholicy z dysbiozą mieli mniejszą medianę liczebności Bacteroidetes i wysoką liczebność Proteobacteria. Badanie nie miało wpływu na osoby, które nie były alkoholikami.
Jednak; jest kilka dobrych wiadomości na temat ograniczania się do alkoholizmu i że może to być korzystne dla bakterii jelitowych. Jeśli umiarkowanie spożywałeś odpowiedzialnie czerwone wino, polifenole w winie może pomóc w poprawie flory jelitowej. Ciesz się kieliszkiem wina raz na jakiś czas jako małą ucztą, której nie należy brać za pewnik.
Niewystarczający sen
W jednym z poprzednich artykułów rozmawialiśmy o tym, jak osiągnąć dobry sen przez zioła. Kiedy w naszym gorączkowym życiu nie zasypiamy ani nie śpimy, wpływa to na nas z powodu różnych problemów zdrowotnych, w tym choroba serca i otyłość, W Badanie 2016, naukowcy odkryli wpływ krótkotrwałej deprywacji snu na mikroflorę jelitową po dwóch dniach.
Kiedy nasze ciało nie otrzymuje zalecanych 8 godzin snu, nasze jelita zbierają ogromne żniwo, ponieważ czujemy się ospali i wyczerpani. Tak więc, aby upewnić się, że nasze mikrobiom jelitowy zostanie pod opieką, zalecamy wyłączenie urządzeń elektronicznych co najmniej 30 minut przed rozpoczęciem snu. Wyłącz wszystkie światła i nie pij żadnych płynów co najmniej dwie godziny przed snem, zamknij oczy i weź głęboki oddech w stanie medytacji i zrelaksuj się, gdy odpływasz w senne miasto.
Niewystarczające ćwiczenia
W naszym dynamicznym stylu życia i stresującej pracy ciężko jest znaleźć czas na ćwiczenia. Ale kiedy faktycznie znajdujemy czas na ćwiczenia, nie tylko nasze umysły czują się dobrze; ale nasze ciało i jelita również dobrze się czują. Jednak rzeczy zawsze pojawiają się, gdy wykonujemy rutynowe ćwiczenia i musimy całkowicie zrezygnować z ćwiczeń. To zdarza się każdemu z nas i ciężko jest nadrobić to, w którym przerwieliśmy, kiedy próbowaliśmy ćwiczyć.
Kiedy nie ćwiczymy co najmniej kilka razy w tygodniu, nasze ciała odbijają się na nas, gdy przybieramy na wadze. stres jest zbyt wysokii mamy większa szansa na przewlekłą chorobę. Kiedy tak się dzieje, nasza flora jelitowa jest ogromną wadą. Tutaj, w klinice, staramy się informować naszych pacjentów o znaczeniu ćwiczeń i że nie tylko zmienia to ich życie, ale także całkowicie zmienia ich nastrój.
Jednak nie należy po prostu wykonywać ciężkiej rutyny ćwiczeń, podczas której można się zranić. Zacznij od treningu o niskiej intensywności, a następnie rozbudowuj go w miarę upływu czasu, ponieważ flora jelitowa Ci za to podziękuje.
Na koniec, tutaj w Injury Medical chcemy Cię informować o żywieniu i sposobach, które pomogą Ci poprawić swoje dolegliwości dzięki niespodziankom 5. Ale także edukować cię na temat tego, co może zranić twoje jelita. Dzięki tym niespodziankom i drobnym zmianom w codziennym życiu twoje jelita będą Ci wdzięczne za długi dystans.
Zasoby NCBI
Według dowodów z badania 2016układ odpornościowy jelit ma zasadnicze znaczenie dla zapobiegania różnym chorobom i często może przyczyniać się do zaburzeń metabolicznych. Jednak może również pomóc w wyznaczeniu celu leczenia w przypadku obserwowania ogólnoustrojowego zapalenia w insulinooporności. Co więcej, zmodyfikowana odporność jelit została powiązana ze zmianami w mikroflorze jelitowej, funkcjonowaniu bariery jelitowej, komórkach odpornościowych w jelitach i oporności na antygeny, które dostają się do przewodu pokarmowego. Chociaż wcześniej uważano, że zwiększa to ryzyko dolegliwości przełyku, w tym infekcji patogennych i przewlekłego zapalenia, które może ostatecznie prowadzić do przewlekłych problemów zdrowotnych.
Ciała ketonowe są tworzone przez wątrobę i wykorzystywane jako źródło energii, gdy glukoza nie jest łatwo dostępna w ludzkim ciele. Dwa główne ciała ketonowe to acetooctan (AcAc) i 3-beta-hydroksymaślan (3HB), podczas gdy aceton jest trzecim i najmniej występującym ciałem ketonowym. Ketony są zawsze obecne we krwi, a ich poziom wzrasta podczas postu i długotrwałych ćwiczeńketogenezy jest biochemicznym procesem, w którym organizmy produkują ciała ketonowe poprzez rozkład kwasów tłuszczowych i ketogennych aminokwasów.
Ciała ketonowe są generowane głównie w mitochondria komórek wątroby. Ketogeneza występuje, gdy poziom glukozy we krwi jest niski, szczególnie po wyczerpaniu innych komórkowych zapasów węglowodanów, takich jak glikogen. Mechanizm ten może również wystąpić, gdy nie ma wystarczającej ilości insuliny. Produkcja ciał ketonowych jest ostatecznie inicjowana w celu udostępnienia energii, która jest magazynowana w ludzkim ciele w postaci kwasów tłuszczowych. Ketogeneza zachodzi w mitochondriach, gdzie jest niezależnie regulowana.
Abstrakcyjny
Metabolizm ciała ketonowego jest centralnym węzłem homeostazy fizjologicznej. W tym przeglądzie omawiamy, w jaki sposób ketony służą dyskretnemu dostrajaniu ról metabolicznych, które optymalizują wydajność narządów i organizmu w zakresie różnych pozostałości składników odżywczych oraz chronią przed zapaleniem i urazami w wielu układach narządów. Tradycyjnie postrzegane jako substraty metaboliczne związane tylko z ograniczeniem węglowodanów, ostatnie obserwacje podkreślają znaczenie ciał ketonowych jako istotnych mediatorów metabolicznych i sygnalizacyjnych w przypadku obfitości węglowodanów. Uzupełniając repertuar znanych opcji terapeutycznych w chorobach układu nerwowego, pojawiły się potencjalne role ciał ketonowych w raku, a także intrygujące role ochronne w sercu i wątrobie, otwierając możliwości terapeutyczne w chorobach związanych z otyłością i układu krążenia. W celu pogodzenia klasycznego dogmatu ze współczesnymi obserwacjami omawiane są kontrowersje związane z metabolizmem i sygnalizacją ketonów.
Wprowadzenie
Ciała ketonowe są istotnym alternatywnym źródłem paliwa metabolicznego dla wszystkich dziedzin życia, eukarii, bakterii i archeonów (Aneja i in., 2002; Cahill GF Jr, 2006; Krishnakumar i in., 2008). Metabolizm ciał ketonowych u ludzi został wykorzystany do napędzania mózgu w okresowych okresach niedoboru składników odżywczych. Ciała ketonowe są przeplatane kluczowymi szlakami metabolicznymi ssaków, takimi jak a-utlenianie (FAO), cykl kwasów trikarboksylowych (TCA), glukoneogeneza, lipogeneza de novo (DNL) i biosynteza steroli. U ssaków ciała ketonowe są wytwarzane głównie w wątrobie z acetylo-CoA pochodzącego z FAO i są transportowane do tkanek pozawątrobowych w celu ostatecznego utlenienia. Ta fizjologia dostarcza alternatywnego paliwa, które jest wzmacniane przez stosunkowo krótkie okresy postu, co zwiększa dostępność kwasów tłuszczowych i zmniejsza dostępność węglowodanów (Cahill GF Jr, 2006; McGarry i Foster, 1980; Robinson i Williamson, 1980). Utlenianie ciała ketonowego staje się znaczącym czynnikiem przyczyniającym się do ogólnego metabolizmu energetycznego ssaków w tkankach pozawątrobowych w niezliczonych stanach fizjologicznych, w tym na czczo, głodzie, okresie noworodkowym, po wysiłku fizycznym, ciąży i przestrzeganiu diety niskowęglowodanowej. Całkowite stężenie ciał ketonowych w krążeniu u zdrowych dorosłych ludzi zwykle wykazuje wahania dobowe w zakresie około 100-250 μM, wzrasta do ~ 1 mM po długotrwałym wysiłku lub 24 godz. Na czczo i może kumulować się nawet do 20 mM w stanach patologicznych, takich jak cukrzycowa kwasica ketonowa ( Cahill GF Jr, 2006; Johnson i in., 1969b; Koeslag i in., 1980; Robinson i Williamson, 1980; Wildenhoff i in., 1974). Ludzka wątroba produkuje do 300 g ciał ketonowych dziennie (Balasse i Fery, 1989), które stanowią od 5% całkowitego wydatku energetycznego w stanach po posiłku, na czczo i z głodem (Balasse i in., 20; Cox i in. al., 1978).
Ostatnie badania podkreślają teraz rolę imperatywną dla ciał ketonowych w metabolizmie komórek ssaków, homeostazie i sygnalizacji w szerokim zakresie stanów fizjologicznych i patologicznych. Oprócz wykorzystywania jako paliwa energetyczne w tkankach pozawątrobowych, takich jak mózg, serce lub mięśnie szkieletowe, ciałka ketonowe odgrywają zasadniczą rolę jako mediatory sygnałowe, czynniki odpowiedzialne za potranslacyjną modyfikację białka (PTM) i modulatory stanu zapalnego i stres oksydacyjny. W tym przeglądzie przedstawiamy klasyczne i współczesne poglądy o plejotropowej roli ciał ketonowych i ich metabolizmu.
Przegląd metabolizmu ciała ketonowego
Tempo ketogenezy wątrobowej jest regulowane przez serię fizjologicznych i biochemicznych przemian tłuszczu. Podstawowe regulatory obejmują lipolizę kwasów tłuszczowych z triacylogliceroli, transport do i przez błonę plazmatyczną hepatocytów, transport do mitochondriów za pośrednictwem palmitoilotransferazy karnityny 1 (CPT1), spiralę P-oksydacji, aktywność cyklu TCA i stężenia pośrednie, potencjał redoks i regulatory hormonalne tych procesów, głównie glukagonu i insuliny [przegląd w (Arias i wsp., 1995; Ayte i wsp., 1993; Ehara i wsp., 2015; Ferre i wsp., 1983; Kahn i wsp., 2005; McGarry i Foster , 1980; Williamson i in., 1969)]. Klasycznie ketogeneza jest postrzegana jako szlak pośredni, w którym acetylo-CoA pochodzący z a-utleniania przewyższa aktywność syntazy cytrynianowej i/lub dostępność szczawiooctanu do kondensacji z wytworzeniem cytrynianu. Trójwęglowe produkty pośrednie wykazują aktywność antyketogenną, prawdopodobnie ze względu na ich zdolność do powiększania puli szczawiooctanów w celu spożycia acetylo-CoA, ale samo stężenie acetylo-CoA w wątrobie nie determinuje szybkości ketogenicznej (Foster, 1967; Rawat i Menahan, 1975; Williamson i in., 1969). Regulacja ketogenezy przez zdarzenia hormonalne, transkrypcyjne i potranslacyjne razem wspierają pogląd, że mechanizmy molekularne, które dostrajają tempo ketogenu, pozostają nie w pełni poznane (patrz Regulacja HMGCS2 i SCOT/OXCT1).
Ketogeneza zachodzi głównie w macierzy mitochondrialnej wątroby z szybkością proporcjonalną do całkowitego utleniania tłuszczu. Po transporcie łańcuchów acylowych przez błony mitochondrialne i p-oksydacji, izoforma mitochondrialna syntazy 3-hydroksymetyloglutarylo-CoA (HMGCS2) katalizuje los powodując kondensację acetoacetylo-CoA (AcAc-CoA) i acetylo-CoA w celu wytworzenia HMG-CoA (Rys. 1A). Liaza HMG-CoA (HMGCL) rozszczepia HMG-CoA, aby uwolnić acetylo-CoA i acetooctan (AcAc), a ten ostatni jest redukowany do d - a - hydroksymaślanu (d-? OHB) przez zależną od fosfatydylocholiny mitochondrialną dehydrogenazę d-aOHB ( BDH1) w reakcji bliskiej równowagi sprzężonej z NAD + / NADH (Bock i Fleischer, 1975; LEHNINGER i wsp., 1960). Stała równowagi BDH1 sprzyja wytwarzaniu d-a-OHB, ale stosunek ciał ketonowych AcAc / d-a-OHB jest wprost proporcjonalny do mitochondrialnego stosunku NAD + / NADH, a zatem aktywność oksydoreduktazy BDH1 moduluje mitochondrialny potencjał redoks (Krebs i wsp., 1969; Williamson i wsp., 1967). AcAc może również spontanicznie dekarboksylować do acetonu (Pedersen, 1929), źródła słodkiego zapachu u ludzi cierpiących na kwasicę ketonową (tj. Całkowita zawartość ciał ketonowych w surowicy> ~ 7 mM; AcAc pKa 3.6, OHB pKa 4.7). Mechanizmy transportu ciał ketonowych przez wewnętrzną błonę mitochondrialną nie są znane, ale AcAc / d-aOHB są uwalniane z komórek za pośrednictwem transporterów monokarboksylanów (u ssaków MCT 1 i 2, znane również jako członkowie rodziny substancji rozpuszczonych 16A 1 i 7) i transportowany w krążeniu do tkanek pozawątrobowych w celu ostatecznego utleniania (Cotter i wsp., 2011; Halestrap i Wilson, 2012; Halestrap, 2012; Hugo i wsp., 2012). Stężenia krążących ciał ketonowych są wyższe niż w tkankach pozawątrobowych (Harrison i Long, 1940), co wskazuje, że ciała ketonowe są transportowane w dół gradientu stężeń. Mutacje powodujące utratę funkcji w MCT1 są związane ze spontanicznymi napadami kwasicy ketonowej, co sugeruje krytyczną rolę w imporcie ciał ketonowych.
� Z wyjątkiem potencjalnego przekierowania ciał ketonowych na nieoksydacyjne losy (patrz Nieoksydacyjne metaboliczne losy ciał ketonowych), hepatocyty nie mają zdolności do metabolizowania wytwarzanych przez nie ciał ketonowych. Ciała ketonowe syntetyzowane de novo przez wątrobę są (i) katabolizowane w mitochondriach tkanek pozawątrobowych do acetylo-CoA, który jest dostępny w cyklu TCA do końcowego utleniania (ryc. 1A), (ii) przekierowany do szlaków lipogenezy lub syntezy steroli ( Ryc. 1B) lub (iii) wydalane z moczem. Jako alternatywne paliwo energetyczne ciała ketonowe są intensywnie utleniane w sercu, mięśniach szkieletowych i mózgu (Balasse i Fery, 1989; Bentourkia i in., 2009; Owen i in., 1967; Reichard i in., 1974; Sultan, 1988 ). Pozawątrobowa mitochondrialna BDH1 katalizuje pierwszą reakcję utleniania -OHB, przekształcając ją w AcAc (LEHNINGER i wsp., 1960; Sandermann i wsp., 1986). Cytoplazmatyczna dehydrogenaza d-AOHB (BDH2) z tylko 20% identycznością sekwencji z BDH1 ma wysoką Km dla ciał ketonowych, a także odgrywa rolę w homeostazie żelaza (Davuluri i wsp., 2016; Guo i wsp., 2006) . W pozawątrobowej macierzy mitochondrialnej AcAc jest aktywowany do AcAc-CoA poprzez wymianę ugrupowania CoA z sukcynylo-CoA w reakcji katalizowanej przez unikalną ssaczą transferazę CoA, transferazę sukcynylo-CoA:3-oksokwas-CoA (SCOT, transferaza CoA; kodowane przez OXCT1), w wyniku reakcji bliskiej równowadze. Energia swobodna uwalniana przez hydrolizę AcAc-CoA jest większa niż energia sukcynylo-CoA, co sprzyja tworzeniu AcAc. Tak więc przepływ oksydacyjny ciała ketonowego następuje w wyniku działania masowego: obfita podaż AcAc i szybkie zużycie acetylo-CoA przez syntazę cytrynianową sprzyja tworzeniu AcAc-CoA (+ bursztynian) przez SCOT. Warto zauważyć, że w przeciwieństwie do glukozy (heksokinazy) i kwasów tłuszczowych (syntetazy acylo-CoA), aktywacja ciał ketonowych (SCOT) do postaci ulegającej utlenieniu nie wymaga inwestycji ATP. Odwracalna reakcja tiolazy AcAc-CoA [katalizowana przez dowolną z czterech mitochondrialnych tiolaz kodowanych przez ACAA2 (kodujący enzym znany jako T1 lub CT), ACAT1 (kodujący T2), HADHA lub HADHB] daje dwie cząsteczki acetylo-CoA, które wchodzą w cykl TCA (Hersh i Jencks, 1967; Stern i in., 1956; Williamson i in., 1971). Podczas stanów ketonowych (tj. całkowita ilość ketonów w surowicy > 500 μM), ciała ketonowe stają się znaczącymi uczestnikami wydatkowania energii i są szybko wykorzystywane w tkankach, aż do wychwytu lub nasycenia utleniania (Balasse i wsp., 1978; Balasse i Fery, 1989 Edmond i wsp., 1987). Bardzo małą część ciał ketonowych pochodzących z wątroby można łatwo zmierzyć w moczu, a wskaźniki wykorzystania i reabsorpcji przez nerki są proporcjonalne do stężenia w krążeniu (Goldstein, 1987; Robinson i Williamson, 1980). Podczas stanów wysoce ketotycznych (> 1 mM w osoczu), ketonuria służy jako półilościowy reporter ketozy, chociaż większość testów klinicznych ciał ketonowych w moczu wykrywa AcAc, ale nie ?OHB (Klocker i wsp., 2013).
Substraty ketogeniczne i ich wpływ na metabolizm hepatocytów
Substancje ketogenne obejmują kwasy tłuszczowe i aminokwasy (ryc. 1B). Katabolizm aminokwasów, w szczególności leucyny, generuje około 4% ciał ketonowych w stanie poabsorpcyjnym (Thomas i wsp., 1982). Tak więc pula substratu acetylo-CoA do generowania ciał ketonowych pochodzi głównie z kwasów tłuszczowych, ponieważ w stanach zmniejszonego dostarczania węglowodanów pirogronian wchodzi do cyklu wątrobowego TCA głównie poprzez anaplerozę, tj. Zależną od ATP karboksylację do szczawiooctanu (OAA) lub do jabłczanu (MAL), a nie oksydacyjnej dekarboksylacji do acetylo-CoA (Jeoung i wsp., 2012, Magnusson i wsp., 1991, Merritt i wsp., 2011). W wątrobie glukoza i pirogronian przyczyniają się w sposób nieistotny do ketogenezy, nawet gdy maksymalna dekarboksylacja pirogronianu do acetylo-CoA jest największa (Jeoung i wsp., 2012).
Acetyl-CoA obejmuje kilka ról stanowiących integralną część wątrobowego metabolizmu pośredniego poza generowaniem ATP poprzez końcowe utlenianie (patrz także: Integracja metabolizmu ciała ketonowego, modyfikacja potranslacyjna i fizjologia komórki). Acetyl-CoA allosterycznie aktywuje (i) karboksylazę pirogronianową (PC), aktywując tym samym mechanizm metabolicznej kontroli, który zwiększa anaplerotyczne wejście metabolitów do cyklu TCA (Owen i wsp., 2002, Scrutton i Utter, 1967) i (ii) dehydrogenazę pirogronianową kinaza, która fosforyluje i hamuje dehydrogenazę pirogronianową (PDH) (Cooper i wsp., 1975), co dodatkowo zwiększa przepływ pirogronianu do cyklu TCA poprzez anapterozę. Ponadto, cytoplazmatyczny acetylo-CoA, którego pula jest wzmacniana przez mechanizmy, które przekształcają mitochondrialny acetylo-CoA w transportowalne metabolity, hamuje utlenianie kwasów tłuszczowych: karboksylaza acetyl-CoA (ACC) katalizuje konwersję acetylo-CoA do malonylo-CoA, lipogennego substratu i allosteryczny inhibitor mitochondrialnego CPT1 [omówionego w (Kahn i wsp., 2005, McGarry and Foster, 1980)]. W ten sposób mitochondrialna pula acetylo-CoA reguluje i jest regulowana przez szlak spillover ketogenezy, który koordynuje kluczowe aspekty wątrobowego pośredniego metabolizmu.
Nieoksydacyjne metaboliczne losy ciał ketonowych
Dominującym losem ketonów pochodzących z wątroby jest pozawątrobowe utlenianie zależne od SCOT. Jednak AcAc może być eksportowany z mitochondriów i wykorzystywany w szlakach anabolicznych poprzez konwersję do AcAc-CoA w reakcji zależnej od ATP katalizowanej przez cytoplazmatyczną syntetazę acetoacetylo-CoA (AACS, Fig. 1B). Szlak ten jest aktywny podczas rozwoju mózgu oraz w okresie laktacji (Morris, 2005; Robinson i Williamson, 1978; Ohgami i in., 2003). AACS jest również silnie wyrażany w tkance tłuszczowej i aktywowanych osteoklastach (Aguilo i wsp., 2010; Yamasaki i wsp., 2016). Cytoplazmatyczny AcAc-CoA może być kierowany przez cytozolowy HMGCS1 w kierunku biosyntezy steroli lub rozszczepiany przez jedną z dwóch tiolaz cytoplazmatycznych do acetylo-CoA (ACAA1 i ACAT2), karboksylowany do malonylo-CoA i przyczyniać się do syntezy kwasów tłuszczowych (Bergstrom et al. wsp., 1984; Edmond, 1974; Endemann i wsp., 1982; Geelen i wsp., 1983; Webber i Edmond, 1977).
Chociaż znaczenie fizjologiczne nie zostało jeszcze ustalone, ketony mogą służyć jako substraty anaboliczne nawet w wątrobie. W sztucznych kontekstach eksperymentalnych AcAc może przyczyniać się do powstania nawet połowy nowo zsyntetyzowanego lipidu i do 75% nowego zsyntetyzowanego cholesterolu (Endemann i wsp., 1982; Geelen i wsp., 1983; Freed i wsp., 1988). Ponieważ AcAc pochodzi z niecałkowitego utleniania tłuszczu w wątrobie, zdolność AcAc do udziału w lipogenezie in vivo sugerowałaby jałowy cykl wątrobowy, w którym ketony pochodzące z tłuszczów mogą być wykorzystywane do produkcji lipidów, co jest pojęciem, którego znaczenie fizjologiczne wymaga walidacji eksperymentalnej, ale może służyć role adaptacyjne lub nieadaptacyjne (Solinas i in., 2015). AcAc chciwie dostarcza cholesterogenezę, z niskim AACS Km-AcAc (~50 ?M) sprzyjającym aktywacji AcAc nawet po posiłku (Bergstrom i wsp., 1984). Zasugerowano dynamiczną rolę cytoplazmatycznego metabolizmu ketonów w pierwotnych neuronach embrionalnych myszy oraz w adipocytach pochodzących z 3T3-L1, ponieważ knockdown AACS zaburza różnicowanie każdego typu komórek (Hasegawa i wsp., 2012a; Hasegawa i wsp., 2012b). Powalenie AACS u myszy in vivo obniżyło poziom cholesterolu w surowicy (Hasegawa i wsp., 2012c). SREBP-2, główny regulator transkrypcji biosyntezy cholesterolu i receptor aktywowany przez proliferatory peroksysomów (PPAR)-? są aktywatorami transkrypcji AACS i regulują jego transkrypcję podczas rozwoju neurytów oraz w wątrobie (Aguilo i wsp., 2010; Hasegawa i wsp., 2012c). Podsumowując, cytoplazmatyczny metabolizm ciał ketonowych może być ważny w wybranych stanach lub naturalnej historii choroby, ale nie jest wystarczający do usuwania ciał ketonowych pochodzących z wątroby, ponieważ masywna hiperketonemia występuje w warunkach selektywnego upośledzenia pierwotnego losu oksydacyjnego poprzez mutacje utraty funkcji SCOT (Berry i in., 2001; Cotter i in., 2011).
Regulacja HMGCS2 i SCOT / OXCT1
Odejście mitochondrialnego od genu kodującego cytozolowy HMGCS nastąpiło na wczesnym etapie ewolucji kręgowców ze względu na potrzebę wspierania ketogenezy wątrobowej u gatunków o wyższym stosunku masy mózgu do masy ciała (Boukaftane i wsp., 1994; Cunnane i Crawford, 2003). Naturalnie występujące mutacje utraty funkcji HMGCS2 u ludzi powodują napady hipoglikemii hipoketotycznej (Pitt i wsp., 2015; Thompson i wsp., 1997). Silna ekspresja HMGCS2 jest ograniczona do hepatocytów i nabłonka okrężnicy, a jego ekspresja i aktywność enzymatyczna są koordynowane przez różne mechanizmy (Mascaro i wsp., 1995; McGarry i Foster, 1980; Robinson i Williamson, 1980). Podczas gdy pełny zakres stanów fizjologicznych wpływających na HMGCS2 wymaga dalszego wyjaśnienia, jego ekspresja i/lub aktywność jest regulowana we wczesnym okresie poporodowym, starzenia się, cukrzycy, głodzie lub przyjmowaniu diety ketogenicznej (Balasse i Fery, 1989; Cahill GF Jr, 2006 Girard i in., 1992; Hegardt, 1999; Satapati i in., 2012; Sengupta i in., 2010). U płodu metylacja regionu flankującego 5� genu Hmgcs2 jest odwrotnie skorelowana z jego transkrypcją i ulega częściowemu odwróceniu po urodzeniu (Arias i wsp., 1995; Ayte i wsp., 1993; Ehara i wsp., 2015; Ferre i wsp., 1983; ., XNUMX). Podobnie, wątrobowe Bdh1 wykazuje rozwojowy wzorzec ekspresji, wzrastający od urodzenia do odsadzenia, a także jest indukowany przez dietę ketogeniczną w sposób zależny od czynnika wzrostu fibroblastów (FGF)-21 (Badman i wsp., 2007; Zhang i wsp., 1989 ). Ketogeneza u ssaków jest wysoce wrażliwa zarówno na insulinę, jak i glukagon, które są odpowiednio tłumione i stymulowane (McGarry i Foster, 1977). Insulina hamuje lipolizę tkanki tłuszczowej, tym samym pozbawiając ketogenezę jej substratu, podczas gdy glukagon zwiększa przepływ ketogenny poprzez bezpośredni wpływ na wątrobę (Hegardt, 1999). Transkrypcja Hmgcs2 jest stymulowana przez przedni czynnik transkrypcyjny FOXA2, który jest hamowany przez kinazę insulinowo-fosfatydyloinozytol-3/Akt i jest indukowany przez sygnalizację glukagon-cAMP-p300 (Arias i in., 1995; Hegardt, 1999; Quant i in. , 1990; Thumelin i wsp. 1993; von Meyenn i wsp. 2013; Wolfrum i wsp. 2004; Wolfrum i wsp. 2003). PPAR? (Rodriguez i wsp., 1994) wraz ze swoim celem, FGF21 (Badman i wsp., 2007) również indukują transkrypcję Hmgcs2 w wątrobie podczas głodzenia lub podawania diety ketogenicznej (Badman i wsp., 2007; Inagaki i wsp., 2007 ). Indukcja PPAR? może wystąpić przed przejściem z fizjologii płodowej do noworodkowej, podczas gdy aktywacja FGF21 może być faworyzowana we wczesnym okresie noworodkowym poprzez hamowanie deacetylazy histonowej (HDAC)-3, w której pośredniczy ?OHB (Rando i wsp., 2016). zależne od mTORC1 (ssaczego celu kompleksu rapamycyny 1) hamowanie PPAR? aktywność transkrypcyjna jest również kluczowym regulatorem ekspresji genu Hmgcs2 (Sengupta i wsp., 2010), a wątrobowy PER2, główny oscylator dobowy, pośrednio reguluje ekspresję Hmgcs2 (Chavan i wsp., 2016). Ostatnie obserwacje wskazują, że interleukina-6 wywołana przez nowotwór pozawątrobowy zaburza ketogenezę poprzez PPAR? tłumienie (Flint i in., 2016).
Aktywność enzymu HMGCS2 reguluje się za pomocą wielu PTM. Fosforylacja seryny HMGCS2 wzmacniała jej aktywność in vitro (Grimsrud i wsp., 2012). Aktywność HMGCS2 jest allosterycznie hamowana sukcynylacją sukcynylo-CoA i lizyny (Arias i wsp., 1995, Hegardt, 1999, Lowe i Tubbs, 1985, Quant i wsp., 1990, Rardin i wsp., 2013, Reed i wsp., 1975; Thumelin i wsp., 1993). Sukcynylacja reszt lizyny HMGCS2, HMGCL i BDH1 w mitochondriach wątrobowych jest celami zależnej od NAD + deacylazy sirtuiny 5 (SIRT5) (Rardin i wsp., 2013). Aktywność HMGCS2 jest również zwiększona przez deacetylację lizyny SIRT3 i możliwe jest, że przesłuch między acetylacją a sukcynylacją reguluje aktywność HMGCS2 (Rardin i wsp., 2013, Shimazu i wsp., 2013). Pomimo zdolności tych PTM do regulowania HMGCS2 Km i Vmax, fluktuacje tych PTM nie zostały jeszcze dokładnie odwzorowane i nie zostały potwierdzone jako mechanistyczne czynniki ketogenezy in vivo.
SCOT ulega ekspresji we wszystkich komórkach ssaków, które zawierają mitochondria, z wyjątkiem komórek hepatocytów. Znaczenie aktywności SCOT i ketolizy wykazano u myszy SCOT-KO, które wykazywały jednolitą śmiertelność z powodu hipoglikemii hiperketonemicznej w ciągu 48 godzin po urodzeniu (Cotter i wsp., 2011). Specyficzna tkankowa utrata SCOT w neuronach lub miocytach szkieletowych wywołuje nieprawidłowości metaboliczne podczas głodu, ale nie jest śmiertelna (Cotter i wsp., 2013b). U ludzi niedobór SCOT objawia się we wczesnym okresie życia ciężką kwasicą ketonową, powodując letarg, wymioty i śpiączkę (Berry i in., 2001; Fukao i in., 2000; Kassovska-Bratinova i in., 1996; Niezen-Koning i in. , 1997; Saudubray i wsp., 1987; Snyderman i wsp., 1998; Tildon i Cornblath, 1972). Stosunkowo niewiele wiadomo na poziomie komórkowym na temat regulatorów ekspresji genu SCOT i białek. Ekspresja mRNA Oxct1 oraz białko i aktywność SCOT są zmniejszone w stanach ketotycznych, prawdopodobnie przez mechanizmy zależne od PPAR (Fenselau i Wallis, 1974; Fenselau i Wallis, 1976; Grinblat i wsp., 1986; Okuda i wsp., 1991; Turko i wsp., 2001; Grinblat i wsp., 2010; ., 1; Wentz i in., 2). W cukrzycowej kwasicy ketonowej niedopasowanie między wątrobową ketogenezą a pozawątrobowym utlenianiem nasila się z powodu upośledzenia aktywności SCOT. Nadekspresja niezależnego od insuliny transportera glukozy (GLUT1/SLC1A2009) w kardiomiocytach również hamuje ekspresję genu Oxct1 i obniża końcowe utlenianie ketonów w stanie nieketotycznym (Yan i wsp., 122). W wątrobie obfitość mRNA Oxct3 jest tłumiona przez mikroRNA-27 i metylację histonów H3K2011me1, które są widoczne podczas przejścia z okresu płodowego do okresu noworodkowego (Thorrez i wsp., 1). Jednak tłumienie ekspresji Oxct1 w wątrobie w okresie poporodowym można przede wszystkim przypisać usunięciu krwiotwórczych komórek progenitorowych z ekspresją Oxct1 z wątroby, a nie utracie wcześniej istniejącej ekspresji Oxct2008 w terminalnie zróżnicowanych hepatocytach. W rzeczywistości ekspresja mRNA OxctXNUMX i białka SCOT w zróżnicowanych hepatocytach jest niezwykle niska (Orii i wsp., XNUMX).
SCOT jest również regulowany przez PTM. Enzym jest hiperacetylowany w mózgach myszy SIRT3 KO, które również wykazują zmniejszoną produkcję acetylo-CoA zależną od AcAc (Dittenhafer-Reed et al., 2015). Nieenzymatyczne nitrowanie reszt tyrozynowych SCOT również osłabia jego aktywność, o czym donoszono w sercach różnych modeli myszy z cukrzycą (Marcondes i wsp., 2001; Turko i wsp., 2001; Wang i wsp., 2010a). W przeciwieństwie do tego azotowanie reszty tryptofanu zwiększa aktywność SCOT (Br g re i in., 2010; Rebrin i in., 2007). Mogą istnieć molekularne mechanizmy nitrowania lub de-nitracji specyficzne dla pozostałości, mające na celu modulowanie aktywności SCOT i wymagają wyjaśnienia.
Kontrowersje w ketogenezie pozawotworowej
U ssaków głównym organem ketogennym jest wątroba i tylko hepatocyty i komórki nabłonka jelita w znacznym stopniu wyrażają mitochondrialną izoformę HMGCS2 (Cotter i wsp., 2013a; Cotter i wsp., 2014; McGarry i Foster, 1980; Robinson i Williamson, 1980) . Beztlenowa fermentacja bakteryjna złożonych polisacharydów daje maślan, który jest absorbowany przez kolonocyty u ssaków w celu ostatecznego utlenienia lub ketogenezy (Cherbuy i wsp., 1995), co może odgrywać rolę w różnicowaniu kolonocytów (Wang i wsp., 2016). Z wyłączeniem komórek nabłonka jelita i hepatocytów, HMGCS2 jest prawie nieobecny w prawie wszystkich innych komórkach ssaków, ale perspektywa pozawątrobowej ketogenezy została zwiększona w komórkach nowotworowych, astrocytach ośrodkowego układu nerwowego, nerkach, trzustce? komórki, nabłonek barwnikowy siatkówki (RPE), a nawet w mięśniach szkieletowych (Adijanto i wsp., 2014; Avogaro i wsp., 1992; El Azzouny i wsp., 2016; Grabacka i wsp., 2016; Kang i wsp., 2015 ; Le Foll i in., 2014; Nonaka i in., 2016; Takagi i in., 2016a; Thevenet i in., 2016; Zhang i in., 2011). Ektopowy HMGCS2 obserwowano w tkankach, które nie mają netto zdolności ketogennej (Cook i in., 2016; Wentz i in., 2010), a HMGCS2 wykazuje prospektywną aktywność `` oświetlenia księżyca '' niezależną od ketogenezy, w tym w jądrze komórkowym (Chen i wsp. , 2016; Kostiuk i in., 2010; Meertens i in., 1998).
Każda tkanka pozawątrobowa, która utlenia ciała ketonowe, ma również potencjał do gromadzenia ciał ketonowych poprzez niezależne mechanizmy HMGCS2 (ryc. 2A). Jednak nie ma tkanki pozawątrobowej, w której stężenie ciał ketonowych w stanie ustalonym przewyższa stężenie w krążeniu (Cotter i in., 2011; Cotter i in., 2013b; Harrison i Long, 1940), podkreślając, że ciała ketonowe są transportowane w dół gradient stężenia poprzez mechanizmy zależne od MCT1 / 2. Jeden mechanizm pozawątrobowej ketogenezy może w rzeczywistości odzwierciedlać względne upośledzenie utleniania ketonów. Dodatkowe potencjalne wyjaśnienia dotyczą dziedziny tworzenia się ciał ketonowych. Po pierwsze, ketogeneza de novo może zachodzić poprzez odwracalną aktywność enzymatyczną tiolazy i SCOT (Weidemann i Krebs, 1969). Gdy stężenie acetylo-CoA jest stosunkowo wysokie, reakcje normalnie odpowiedzialne za utlenianie AcAc działają w odwrotnym kierunku (GOLDMAN, 1954). Drugi mechanizm występuje, gdy związki pośrednie pochodzące z β-utleniania gromadzą się z powodu wąskiego gardła cyklu TCA, AcAc-CoA przekształca się w l-a-OHB-CoA w wyniku reakcji katalizowanej przez mitochondrialną dehydrogenazę 3-hydroksyacylo-CoA, a następnie przez 3-hydroksybutyryl Deacylazę CoA do l-aOHB, której nie można odróżnić metodą spektrometrii mas lub spektroskopii rezonansowej od fizjologicznego enancjomeru d-aOHB (Reed i Ozand, 1980). I-aOHB można odróżnić chromatograficznie lub enzymatycznie od d-aOHB i występuje w tkankach pozawątrobowych, ale nie w wątrobie ani we krwi (Hsu i wsp., 2011). Ketogeneza wątrobowa wytwarza tylko d-aOHB, jedyny enancjomer będący substratem BDH (Ito i wsp., 1984; Lincoln i wsp., 1987; Reed i Ozand, 1980; Scofield i wsp., 1982; Scofield i wsp., 1982). Trzeci mechanizm niezależny od HMGCS2 generuje d-aOHB poprzez katabolizm aminokwasów, szczególnie leucyny i lizyny. Czwarty mechanizm jest pozorny tylko dlatego, że jest spowodowany artefaktem znakowania i dlatego jest nazywany pseudoketogenezą. Zjawisko to można przypisać odwracalności reakcji SCOT i tiolazy i może powodować przeszacowanie obrotu ciała ketonowego z powodu izotopowego rozcieńczenia ciała ketonowego znacznika w tkance pozawątrobowej (Des Rosiers i wsp., 1990; Fink i wsp., 1988) . Niemniej jednak pseudoketogeneza może być pomijalna w większości kontekstów (Bailey i in., 1990; Keller i in., 1978). Schemat (ryc. 2A) wskazuje przydatne podejście do zastosowania, biorąc pod uwagę podwyższone stężenie ketonów w tkankach w stanie stacjonarnym.
� Ostatnio zwrócono uwagę na nerkę jako potencjalnie ketogenny narząd. W ogromnej większości stanów nerka jest konsumentem netto ciał ketonowych pochodzenia wątrobowego, wydalających lub ponownie wchłaniających ciała ketonowe z krwioobiegu, a nerki na ogół nie są generatorem ani koncentratorem ciał ketonowych netto (Robinson i Williamson, 1980). Autorzy klasycznego badania doszli do wniosku, że minimalna ketogeneza nerkowa określona ilościowo w sztucznym układzie doświadczalnym nie była fizjologicznie istotna (Weidemann i Krebs, 1969). Ostatnio w mysich modelach z cukrzycą i niedoborem autofagii wywnioskowano ketogenezę nerkową, ale bardziej prawdopodobne jest, że wielonarządowe przesunięcia w homeostazie metabolicznej zmieniają integracyjny metabolizm ketonów poprzez oddziaływanie na wiele narządów (Takagi i wsp., 2016a; Takagi i wsp., 2016b; Zhang i in., 2011). Jedna z niedawnych publikacji sugerowała ketogenezę nerkową jako mechanizm ochronny przed uszkodzeniem niedokrwienno-reperfuzyjnym w nerkach (Tran i wsp., 2016). Bezwzględne stężenia αOHB w stanie stacjonarnym z ekstraktów tkanki nerki myszy odnotowano przy ~4 mM. Aby sprawdzić, czy jest to wykonalne, obliczyliśmy ilościowo stężenia αOHB w ekstraktach nerkowych myszy karmionych i głodzonych 12 godziny na dobę. Stężenie ?OHB w surowicy wzrosło z ~24 μM do 100 mM po 2 godzinach na czczo (ryc. 24B), podczas gdy stężenia αOHB w stanie stacjonarnym w nerkach około 2 μM po posiłku i tylko 100 mM po 1 godzinach na czczo (ryc. 24C�E), obserwacje zgodne ze stężeniami obliczonymi ponad 2 lat temu (Hems i Brosnan, 45). Pozostaje możliwe, że w stanach ketonowych ciała ketonowe pochodzenia wątrobowego mogą działać renoprotekcyjnie, ale dowody na ketogenezę nerek wymagają dalszego uzasadnienia. Przekonujące dowody potwierdzające prawdziwą ketogenezę pozawątrobową przedstawiono w RPE (Adijanto i wsp., 1970). Zasugerowano, że ta intrygująca przemiana metaboliczna potencjalnie umożliwi ketonom pochodzącym z RPE przepływ do fotoreceptorów lub komórek gleju Mellera, co może pomóc w regeneracji zewnętrznego segmentu fotoreceptora.
„OHB jako mediator sygnalizujący”
Chociaż są one bogate energetycznie, ciała ketonowe pełnią prowokacyjne "niekanoniczne" role sygnalizacyjne w homeostazie komórkowej (ryc. 3) (Newman i Verdin, 2014; Rojas-Morales i in., 2016). Na przykład, aOHB hamuje HDAC klasy I, co zwiększa acetylację histonów i tym samym indukuje ekspresję genów, które ograniczają stres oksydacyjny (Shimazu i wsp., 2013). ?OHB sam w sobie jest kowalencyjnym modyfikatorem histonów w resztach lizyny w wątrobach myszy z cukrzycą na czczo lub myszy z cukrzycą indukowaną streptozotocyną (Xie i wsp., 2016) (patrz również poniżej, Integracja metabolizmu ciał ketonowych, modyfikacja potranslacyjna i fizjologia komórki, oraz Ciała ketonowe, stres oksydacyjny i neuroprotekcja).
?OHB jest również efektorem poprzez receptory sprzężone z białkiem G. Poprzez niejasne mechanizmy molekularne hamuje aktywność współczulnego układu nerwowego i zmniejsza całkowite zużycie energii i tętno poprzez hamowanie sygnalizacji krótkołańcuchowych kwasów tłuszczowych przez receptor 41 sprzężony z białkiem G (GPR41) (Kimura i wsp., 2011). Jeden z najlepiej zbadanych efektów sygnalizacyjnych aOHB zachodzi przez GPR109A (znany również jako HCAR2), członka podrodziny kwasu hydrokarboksylowego GPCR ulegającego ekspresji w tkance tłuszczowej (białej i brązowej) (Tunaru i wsp., 2003) oraz w komórki odpornościowe (Ahmed i wsp., 2009). AOHB jest jedynym znanym endogennym ligandem receptora GPR109A (EC50 ~770 µM) aktywowanym przez d-AOHB, l-AOHB i maślan, ale nie AcAc (Taggart i wsp., 2005). Wysoki próg stężenia dla aktywacji GPR109A osiąga się poprzez przestrzeganie diety ketogenicznej, głód lub podczas kwasicy ketonowej, co prowadzi do zahamowania lipolizy tkanki tłuszczowej. Działanie antylipolityczne GPR109A przebiega poprzez hamowanie cyklazy adenylylowej i zmniejszenie cAMP, hamując wrażliwą na hormony lipazę triglicerydową (Ahmed i wsp., 2009; Tunaru i wsp., 2003). Tworzy to pętlę ujemnego sprzężenia zwrotnego, w której ketoza hamuje modulację ketogenezy poprzez zmniejszenie uwalniania niezestryfikowanych kwasów tłuszczowych z adipocytów (Ahmed et al., 2009; Taggart et al., 2005), efekt, który można zrównoważyć przez napęd współczulny, który stymuluje lipolizę. Niacyna (witamina B3, kwas nikotynowy) jest silnym (EC50 ~ 0.1 M) ligandem dla GRP109A, skutecznie stosowanym od dziesięcioleci w dyslipidemii (Benyo i wsp., 2005; Benyo i wsp., 2006; Fabbrini i wsp., 2010a; Lukasova i wsp., 2011; Tunaru i wsp., 2003). Podczas gdy niacyna wzmaga odwrócony transport cholesterolu w makrofagach i zmniejsza zmiany miażdżycowe (Lukasova i wsp., 2011), wpływ ?OHB na zmiany miażdżycowe pozostaje nieznany. Chociaż receptor GPR109A pełni role ochronne i istnieją intrygujące powiązania między stosowaniem diety ketogenicznej w udarze a chorobami neurodegeneracyjnymi (Fu i wsp., 2015; Rahman i wsp., 2014), ochronnej roli aOHB za pośrednictwem GPR109A nie wykazano in vivo .
Wreszcie, ?OHB może wpływać na apetyt i sytość. Metaanaliza badań, w których mierzono wpływ diet ketogenicznych i bardzo niskoenergetycznych, wykazała, że uczestnicy spożywający te diety wykazują większe uczucie sytości w porównaniu z dietami kontrolnymi (Gibson i in., 2015). Jednak prawdopodobnym wyjaśnieniem tego efektu są dodatkowe elementy metaboliczne lub hormonalne, które mogą modulować apetyt. Na przykład, myszy utrzymywane na diecie ketogenicznej dla gryzoni wykazywały zwiększone zużycie energii w porównaniu z myszami karmionymi karmą kontrolną, pomimo podobnego spożycia kalorii, a krążąca leptyna lub geny peptydów regulujących zachowanie żywieniowe nie uległy zmianie (Kennedy i wsp., 2007). Wśród proponowanych mechanizmów sugerujących tłumienie apetytu przez ?OHB znajdują się zarówno sygnalizacja, jak i utlenianie (Laeger et al., 2010). Specyficzna dla hepatocytów delecja genu rytmu okołodobowego (Per2) i badania immunoprecypitacji chromatyny wykazały, że PER2 bezpośrednio aktywuje gen Cpt1a i pośrednio reguluje Hmgcs2, prowadząc do upośledzenia ketozy u myszy z nokautem Per2 (Chavan i wsp., 2016). Myszy te wykazywały upośledzone przewidywanie pokarmu, które zostało częściowo przywrócone przez ogólnoustrojowe podawanie ?OHB. Potrzebne będą dalsze badania, aby potwierdzić, że ośrodkowy układ nerwowy jest bezpośrednim celem ?OHB i czy do zaobserwowanych efektów wymagane jest utlenianie ketonów, czy też zaangażowany jest inny mechanizm sygnalizacji. Inni badacze powoływali się na możliwość miejscowej ketogenezy pochodzącej z astrocytów w brzuszno-przyśrodkowym podwzgórzu jako regulatora przyjmowania pokarmu, ale te wstępne obserwacje również skorzystają na ocenach genetycznych i opartych na przepływie (Le Foll i wsp., 2014). Związek między ketozą a niedoborem składników odżywczych pozostaje interesujący, ponieważ głód i sytość są ważnymi elementami nieudanych prób odchudzania.
Integracja metabolizmu ciała ketonowego, modyfikacji potranslacyjnej i fizjologii komórki
Ciała ketonowe przyczyniają się do skompartmentalizowanej puli acetylo-CoA, kluczowego związku pośredniego, który odgrywa znaczącą rolę w metabolizmie komórkowym (Pietrocola i wsp., 2015). Jedną z funkcji acetylo-CoA jest służenie jako substrat do acetylacji, katalizowanej enzymatycznie modyfikacji kowalencyjnej histonu (Choudhary i wsp., 2014; Dutta i wsp., 2016; Fan i wsp., 2015; Menzies i wsp., 2016 ). Duża liczba dynamicznie acetylowanych białek mitochondrialnych, z których wiele może powstać w wyniku mechanizmów nieenzymatycznych, również wyłoniła się z badań proteomiki obliczeniowej (Dittenhafer-Reed i wsp., 2015; Hebert i wsp., 2013; Rardin i wsp., 2013 ; Shimazu i wsp., 2010). Deacetylazy lizynowe wykorzystują kofaktor cynkowy (np. nukleocytozolowe HDAC) lub NAD+ jako kosubstrat (sirtuiny, SIRT) (Choudhary i wsp., 2014; Menzies i wsp., 2016). Acetyloproteom służy zarówno jako czujnik, jak i efektor całkowitej komórkowej puli acetylo-CoA, ponieważ każda z manipulacji fizjologicznych i genetycznych powoduje nieenzymatyczne globalne zmiany acetylacji (Weinert et al., 2014). Ponieważ metabolity wewnątrzkomórkowe służą jako modulatory acetylacji reszt lizynowych, ważne jest rozważenie roli ciał ketonowych, których liczebność jest wysoce dynamiczna.
• OHB jest epigenetycznym modyfikatorem działającym na co najmniej dwa mechanizmy. Zwiększone poziomy OHB wywołane postem, ograniczeniem kalorii, bezpośrednim podawaniem lub długotrwałym wysiłkiem fizycznym wywołują hamowanie HDAC lub aktywację acetylotransferazy histonów (Marosi i wsp., 2016; Sleiman i wsp., 2016) lub stres oksydacyjny (Shimazu i wsp., 2013) . • Hamowanie HDAC3 przez OHB może regulować fizjologię metaboliczną noworodka (Rando i wsp., 2016). Niezależnie, sam aOHB bezpośrednio modyfikuje reszty lizyny histonu (Xie i wsp., 2016). Długotrwałe głodzenie lub cukrzycowa kwasica ketonowa wywołana steptozotocyną zwiększały a-hydroksybutyrylację histonu. Chociaż liczba miejsc a-hydroksybutyrylowania lizyny i miejsc acetylacji była porównywalna, zaobserwowano stechiometrycznie większe a-hydroksybutyrylowanie histonu niż acetylowanie. P-hydroksybutyrylacja histonu lizyny w porównaniu z acetylacją lub metylacją miała wpływ na różne geny, co sugeruje odmienne funkcje komórkowe. Nie wiadomo, czy a-hydroksybutyrylacja jest spontaniczna czy enzymatyczna, ale rozszerza zakres mechanizmów poprzez ciałka ketonowe dynamicznie wpływające na transkrypcję.
Niezbędne zdarzenia przeprogramowania komórek podczas restrykcji kalorycznej i niedoboru składników odżywczych mogą zachodzić w zależnej od SIRT3 i SIRT5 deacetylacji i desuccynylacji mitochondriów, odpowiednio, regulując białka ketogenne i ketolityczne na poziomie potranslacyjnym w wątrobie i tkankach pozawątrobowych (Dittenhafer-Reed et al., 2015; Hebert i in., 2013; Rardin i in., 2013; Shimazu i in., 2010). Chociaż porównanie stechiometryczne zajętych miejsc niekoniecznie wiąże się bezpośrednio ze zmianami w przepływie metabolicznym, acetylacja mitochondrialna jest dynamiczna i może być napędzana przez stężenie acetylo-CoA lub pH mitochondrialne, a nie enzymatyczne acetylotransferazy (Wagner i Payne, 2013). Fakt, że SIRT3 i SIRT5 modulują aktywność enzymów metabolizujących ciało ketonowe, nasuwa pytanie o wzajemną rolę ketonów w rzeźbieniu acetylproteomu, sukcynyloproteomu i innych dynamicznych celów komórkowych. Rzeczywiście, ponieważ zmiany ketogenezy odzwierciedlają stężenia NAD +, produkcja i obfitość ketonów mogą regulować aktywność sirtuiny, wpływając w ten sposób na całkowite pule acetylo-CoA / sukcynylo-CoA, acylproteom, a tym samym fizjologię mitochondrialną i komórkową. P-hydroksybutyrylacja reszt enzymu lizyny może dodać kolejną warstwę do przeprogramowania komórkowego. W tkankach pozawątrobowych utlenianie ciał ketonowych może stymulować analogiczne zmiany w homeostazie komórkowej. Podczas gdy podział puli acetylo-CoA jest silnie regulowany i koordynuje szerokie spektrum zmian komórkowych, zdolność ciał ketonowych do bezpośredniego kształtowania zarówno mitochondrialnych, jak i cytoplazmatycznych stężeń acetylo-CoA wymaga wyjaśnienia (Chen et al., 2012; Corbet et al., 2016; Pougovkina i in., 2014; Schwer i in., 2009; Wellen i Thompson, 2012). Ponieważ stężenia acetylo-CoA są ściśle regulowane, a acetylo-CoA jest nieprzepuszczalny dla błony, ważne jest, aby wziąć pod uwagę mechanizmy napędowe koordynujące homeostazę acetylo-CoA, w tym szybkość produkcji i końcowego utleniania w cyklu TCA, konwersja do ciał ketonowych, mitochondrialne wypływ przez acetylotransferazę karnityny (CrAT) lub eksport acetylo-CoA do cytozolu po konwersji do cytrynianu i uwolnieniu przez liazę cytrynianową ATP (ACLY). Kluczowe role tych ostatnich mechanizmów w acetylproteomie komórkowym i homeostazie wymagają odpowiedniego zrozumienia roli ketogenezy i utleniania ketonów (Das i wsp., 2015; McDonnell i wsp., 2016; Moussaieff i wsp., 2015; Overmyer i wsp., 2015; Seiler i in., 2014; Seiler i in., 2015; Wellen i in., 2009; Wellen i Thompson, 2012). Aby określić cele i wyniki, potrzebne będą konwergentne technologie w metabolomice i acylopoteomice w tworzeniu modeli modyfikowanych genetycznie.
Przeciwzapalne i przeciwzapalne reakcje na ciała ketonowe
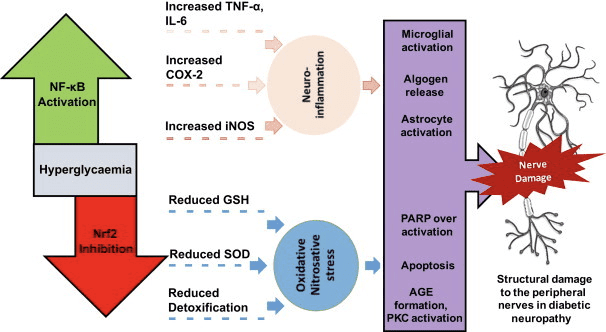
Ketoza i ciała ketonowe modulują stan zapalny i funkcję komórek odpornościowych, ale zaproponowano różne, a nawet rozbieżne mechanizmy. Długotrwała deprywacja składników odżywczych zmniejsza stan zapalny (Youm i in., 2015), ale przewlekła ketoza cukrzycy typu 1 jest stanem prozapalnym (Jain i wsp., 2002; Kanikarla-Marie i Jain, 2015; Kurepa i wsp., 2012 ). Role sygnalizacyjne oparte na mechanizmie dla OHB w zapaleniu pojawiają się, ponieważ wiele komórek układu odpornościowego, w tym makrofagi lub monocyty, obficie eksprymuje GPR109A. Podczas gdy OHB wywiera głównie odpowiedź przeciwzapalną (Fu i wsp., 2014; Gambhir i wsp., 2012; Rahman i wsp., 2014; Youm i wsp., 2015), wysokie stężenia ciał ketonowych, zwłaszcza AcAc, mogą wywołać reakcję prozapalną (Jain i in., 2002; Kanikarla-Marie i Jain, 2015; Kurepa i in., 2012).
Dokonano przeglądu przeciwzapalnej roli ligandów GPR109A w miażdżycy, otyłości, zapalnej chorobie jelit, chorobie neurologicznej i raku (Graff et al., 2016). Ekspresja GPR109A jest zwiększona w komórkach RPE modeli cukrzycowych, ludzi z cukrzycą (Gambhir et al., 2012) oraz w mikrogleju podczas neurodegeneracji (Fu et al., 2014). Efekty przeciwzapalne aOHB są wzmacniane przez nadekspresję GPR109A w komórkach RPE i niwelowane przez farmakologiczne hamowanie lub genetyczny knockout GPR109A (Gambhir i wsp., 2012). ?OHB i egzogenny kwas nikotynowy (Taggart i wsp., 2005), oba nadają działanie przeciwzapalne w TNF? lub stan zapalny wywołany przez LPS przez zmniejszenie poziomu białek prozapalnych (iNOS, COX-2) lub wydzielanych cytokin (TNFa, IL-1a, IL-6, CCL2/MCP-1), częściowo poprzez hamowanie NF Translokacja -?B (Fu i wsp., 2014; Gambhir i wsp., 2012). ?OHB zmniejsza stres ER i inflamasom NLRP3, aktywując odpowiedź na stres antyoksydacyjny (Bae i wsp., 2016; Youm i wsp., 2015). Jednak w zapaleniu neurodegeneracyjnym, ochrona zależna od GPR109A, w której pośredniczy ?OHB, nie obejmuje mediatorów zapalnych, takich jak sygnalizacja szlaku MAPK (np. ERK, JNK, p38) (Fu i wsp., 2014), ale może wymagać PGD1 zależnej od COX-2 produkcja (Rahman i in., 2014). Intrygujące jest to, że makrofag GPR109A jest wymagany do wywierania działania neuroprotekcyjnego w modelu udaru niedokrwiennego (Rahman et al., 2014), ale zdolność ?OHB do hamowania inflamasomu NLRP3 w makrofagach pochodzących ze szpiku kostnego jest niezależna od GPR109A (Youm et al. ., 2015). Chociaż większość badań łączy ?OHB z działaniem przeciwzapalnym, ?OHB może mieć działanie prozapalne i zwiększać markery peroksydacji lipidów w hepatocytach cielęcych (Shi i wsp., 2014). Efekty przeciwzapalne lub prozapalne aOHB mogą zatem zależeć od typu komórki, stężenia aOHB, czasu ekspozycji oraz obecności lub braku komodulatorów.
W przeciwieństwie do OHB, AcAc może aktywować sygnalizację prozapalną. Podwyższony AcAc, szczególnie przy wysokim stężeniu glukozy, nasila uszkodzenie komórek śródbłonka poprzez mechanizm zależny od oksydazy NADPH / stresu oksydacyjnego (Kanikarla-Marie i Jain, 2015). Wysokie stężenia AcAc w pępowinie matek z cukrzycą były skorelowane z wyższym tempem utleniania białek i stężeniem MCP-1 (Kurepa i in., 2012). Wysoki poziom AcAc u pacjentów z cukrzycą był skorelowany z TNF? ekspresja (Jain i wsp., 2002) i AcAc, ale nie OHB, indukowały TNFa, ekspresję MCP-1, akumulację ROS i obniżony poziom cAMP w ludzkich komórkach monocytów U937 (Jain i wsp., 2002; Kurepa i wsp. ., 2012).
Zjawiska sygnalizacyjne zależne od ciał ketonowych są często wyzwalane jedynie przy wysokich stężeniach ciał ketonowych (> 5 mM), aw przypadku wielu badań łączących ketony z działaniem pro lub przeciwzapalnym, poprzez niejasne mechanizmy. Ponadto, ze względu na sprzeczny wpływ OHB w porównaniu z AcAc na stan zapalny oraz zdolność stosunku AcAc / OHB do wpływania na mitochondrialny potencjał redoks, najlepsze eksperymenty oceniające rolę ciał ketonowych w fenotypach komórkowych porównują wpływ AcAc i? OHB w różnych stosunkach i przy różnych skumulowanych stężeniach [np. (Saito i in., 2016)]. Wreszcie AcAc można kupić na rynku tylko jako sól litu lub jako ester etylowy, który przed użyciem wymaga hydrolizy zasadowej. Kation litu niezależnie indukuje kaskady transdukcji sygnału (Manji i wsp., 1995), a anion AcAc jest labilny. Wreszcie, badania z użyciem racemicznego d / l-a-OHB mogą być skomplikowane, ponieważ tylko stereoizomer d-a-OHB może być utleniony do AcAc, ale każdy z d-a-OHB i l-a-OHB może sygnalizować przez GPR109A, hamować inflamasom NLRP3, i służą jako substraty lipogenne.
Ketone Bodies, Oxidative Stress i Neuroprotection
Stres oksydacyjny jest zwykle definiowany jako stan, w którym ROS występują w nadmiarze z powodu nadmiernej produkcji i/lub upośledzonej eliminacji. Rola ciał ketonowych jako przeciwutleniająca i łagodząca stres oksydacyjny została szeroko opisana zarówno in vitro, jak i in vivo, szczególnie w kontekście neuroprotekcji. Ponieważ większość neuronów nie wytwarza skutecznie fosforanów o wysokiej energii z kwasów tłuszczowych, ale utlenia ciała ketonowe, gdy brakuje węglowodanów, neuroprotekcyjne działanie ciał ketonowych jest szczególnie ważne (Cahill GF Jr, 2006; Edmond i in., 1987; Yang i in., 1987). W modelach stresu oksydacyjnego indukcja BDH1 i supresja SCOT sugerują, że metabolizm ciała ketonowego można przeprogramować, aby podtrzymać różne sygnalizacje komórkowe, potencjał redoks lub wymagania metaboliczne (Nagao i wsp., 2016; Tieu i wsp., 2003).
Ciała ketonowe zmniejszają stopień uszkodzenia komórek, urazów, śmierci i niższej apoptozy w neuronach i kardiomiocytach (Haces i wsp., 2008; Maalouf i wsp., 2007; Nagao i wsp., 2016; Tieu i wsp., 2003). Wywoływane mechanizmy są zróżnicowane i nie zawsze są liniowo powiązane z koncentracją. Niskie milimolowe stężenia (d lub l) - A OHB wychwytujący RFT (anion hydroksylowy), podczas gdy AcAc zmiata wiele gatunków RFT, ale tylko w stężeniach przekraczających fizjologiczny zakres (IC50 20 mM) (Haces i in., 67) . I odwrotnie, korzystny wpływ na potencjał redoks łańcucha transportu elektronów jest mechanizmem powszechnie powiązanym z d-aOHB. Podczas gdy wszystkie trzy ciała ketonowe (d / l-aOHB i AcAc) zmniejszyły śmierć komórek neuronalnych i akumulację RFT wyzwalane przez chemiczne hamowanie glikolizy, tylko d-a-OHB i AcAc zapobiegały spadkowi neuronalnego ATP. Odwrotnie, w modelu hipoglikemii in vivo, (d lub l) -? OHB, ale nie AcAc zapobiegał peroksydacji lipidów w hipokampie (Haces i in., 2008; Maalouf i in., 2008; Marosi i in., 2007; Murphy, 2016 ; Tieu i in., 2009). Badania in vivo myszy karmionych dietą ketogenną (2003% tłuszczu kcal i 87% białka) wykazały neuroanatomiczne zróżnicowanie zdolności antyoksydacyjnej (Ziegler et al., 13), gdzie najgłębsze zmiany obserwowano w hipokampie, ze wzrostem peroksydazy glutationowej i całkowitej zdolności przeciwutleniające.
Dieta ketogeniczna, estry ketonowe (patrz także Terapeutyczne zastosowanie diety ketogenicznej i egzogennych ciał ketonowych) lub podawanie OHB wywiera działanie neuroprotekcyjne w modelach udaru niedokrwiennego (Rahman i wsp., 2014); Choroba Parkinsona (Tieu i wsp., 2003); napad toksyczności tlenowej ośrodkowego układu nerwowego (D'Agostino i wsp., 2013); skurcze epileptyczne (Yum i wsp., 2015); encefalomiopatia mitochondrialna, zespół kwasicy mleczanowej i epizodów podobnych do udaru mózgu (MELAS) (Frey i wsp., 2016) oraz choroba Alzheimera (Cunnane i Crawford, 2003; Yin i wsp., 2016). Z kolei niedawny raport wykazał histopatologiczne dowody na progresję neurodegeneracyjną spowodowaną dietą ketogenną w transgenicznym mysim modelu nieprawidłowej naprawy mitochondrialnego DNA, pomimo wzrostu biogenezy mitochondrialnej i sygnatur antyoksydacyjnych (Lauritzen i wsp., 2016). Inne sprzeczne raporty sugerują, że ekspozycja na wysokie stężenia ciał ketonowych wywołuje stres oksydacyjny. Wysokie dawki OHB lub AcAc indukowały wydzielanie tlenku azotu, peroksydację lipidów, zmniejszoną ekspresję SOD, peroksydazy glutationowej i katalazy w hepatocytach cielęcych, podczas gdy w hepatocytach szczurów indukcję szlaku MAPK przypisywano AcAc, ale nie OHB (Abdelmegeed et al., 2004 ; Shi i in., 2014; Shi i in., 2016).
Podsumowując, większość doniesień łączy OHB z osłabieniem stresu oksydacyjnego, ponieważ jego podawanie hamuje wytwarzanie ROS / nadtlenków, zapobiega peroksydacji lipidów i utlenianiu białek, zwiększa poziom białek przeciwutleniających oraz poprawia oddychanie mitochondrialne i produkcję ATP (Abdelmegeed i wsp., 2004; Haces i wsp., 2008; Jain i wsp., 1998; Jain i wsp., 2002; Kanikarla-Marie i Jain, 2015; Maalouf i wsp., 2007; Maalouf i Rho, 2008; Marosi i wsp., 2016; Tieu i in., 2003; Yin i in., 2016; Ziegler i in., 2003). Chociaż AcAc był bardziej bezpośrednio skorelowany niż AOHB z indukcją stresu oksydacyjnego, efekty te nie zawsze są łatwo oddzielone od potencjalnych odpowiedzi prozapalnych (Jain i wsp., 2002; Kanikarla-Marie i Jain, 2015; Kanikarla-Marie i Jain, 2016). Ponadto należy wziąć pod uwagę, że pozorna korzyść antyoksydacyjna zapewniana przez plejotropową dietę ketogenną może nie być transdukowana przez same ciała ketonowe, a neuroprotekcja zapewniana przez ciałka ketonowe może nie być całkowicie przypisana stresowi oksydacyjnemu. Na przykład podczas deprywacji glukozy, w modelu deprywacji glukozy w neuronach korowych, aOHB stymulował przepływ autofagiczny i zapobiegał akumulacji autofagosomów, co było związane ze zmniejszeniem śmierci neuronów (Camberos-Luna i in., 2016). d-aOHB indukuje również kanoniczne białka przeciwutleniające FOXO3a, SOD, MnSOD i katalazę, prospektywnie poprzez hamowanie HDAC (Nagao i wsp., 2016; Shimazu i wsp., 2013).
Niealkoholowa stłuszczenie wątroby (NAFLD) i metabolizm ciała ketonowego
NAFLD związane z otyłością i niealkoholowe stłuszczeniowe zapalenie wątroby (NASH) są najczęstszymi przyczynami chorób wątroby w krajach zachodnich (Rinella i Sanyal, 2016), a niewydolność wątroby wywołana przez NASH jest jedną z najczęstszych przyczyn przeszczepu wątroby. Podczas gdy samo nadmierne przechowywanie triacylogliceroli w hepatocytach >5% masy wątroby (NAFL) nie powoduje degeneracyjnej czynności wątroby, progresja do NAFLD u ludzi koreluje z ogólnoustrojową opornością na insulinę i zwiększonym ryzykiem cukrzycy typu 2 i może przyczyniać się do patogenezy choroba sercowo-naczyniowa i przewlekła choroba nerek (Fabbrini i wsp., 2009; Targher i wsp., 2010; Targher i Byrne, 2013). Mechanizmy patogenne NAFLD i NASH nie są w pełni poznane, ale obejmują nieprawidłowości w metabolizmie hepatocytów, autofagię hepatocytów i stres retikulum endoplazmatycznego, funkcję wątrobowych komórek odpornościowych, zapalenie tkanki tłuszczowej i ogólnoustrojowe mediatory zapalne (Fabbrini i wsp., 2009; Masuoka i Chalasani, 2013 Targher i wsp., 2010; Yang i wsp., 2010). Zaburzenia metabolizmu węglowodanów, lipidów i aminokwasów występują i przyczyniają się do otyłości, cukrzycy i NAFLD u ludzi i organizmów modelowych [przegląd w (Farese i in., 2012; Lin i Accili, 2011; Newgard, 2012; Samuel i Shulman, 2012; Sun i Lazar, 2013)]. Podczas gdy nieprawidłowości hepatocytów w cytoplazmatycznym metabolizmie lipidów są powszechnie obserwowane w NAFLD (Fabbrini i wsp., 2010b), rola metabolizmu mitochondrialnego, który reguluje oksydacyjną utylizację tłuszczów, jest mniej jasna w patogenezie NAFLD. Nieprawidłowości metabolizmu mitochondrialnego występują i przyczyniają się do patogenezy NAFLD/NASH (Hyotylainen i wsp., 2016; Serviddio i wsp., 2011; Serviddio i wsp., 2008; Wei i wsp., 2008). Są ogólne (Felig et al., 1974; Iozzo et al., 2010; Koliaki et al., 2015; Satapati et al., 2015; Satapati et al., 2012; Sunny et al., 2011), ale nie jednolite ( Koliaki i Roden, 2013; Perry i wsp., 2016; Rector i wsp., 2010) zgadzają się, że przed rozwojem NASH w dobrej wierze, wątrobowe utlenianie mitochondriów, a w szczególności utlenianie tłuszczów, nasila się w otyłości, ogólnoustrojowej oporności na insulinę i NAFLD. Jest prawdopodobne, że wraz z postępem NAFLD pojawia się heterogenność zdolności oksydacyjnej, nawet wśród poszczególnych mitochondriów, i ostatecznie funkcja oksydacyjna zostaje osłabiona (Koliaki i in., 2015; Rector i in., 2010; Satapati i in., 2008; Satapati i in., 2012; ., XNUMX).
Ketogeneza jest często używana jako zastępca utleniania tłuszczu w wątrobie. Zaburzenia ketogenezy pojawiają się wraz z postępem NAFLD w modelach zwierzęcych i prawdopodobnie u ludzi. Poprzez nie do końca zdefiniowane mechanizmy hiperinsulinemia hamuje ketogenezę, prawdopodobnie przyczyniając się do hipoketonemii w porównaniu do szczupłych grup kontrolnych (Bergman i wsp., 2007; Bickerton i wsp., 2008; Satapati i wsp., 2012; Soeters i wsp., 2009; Sunny i wsp., 2011; Bickerton i wsp., 2005; Satapati i wsp., 2015; Soeters i wsp., 2001; Sunny i wsp. , 2012; Vice i in., 2010). Niemniej jednak zdolność krążących ciał ketonowych do przewidywania NAFLD jest kontrowersyjna (M nnist i in., 2008; Sanyal i in., 2011). Solidne ilościowe metody spektroskopowe rezonansu magnetycznego w modelach zwierzęcych ujawniły zwiększoną szybkość obrotu ketonowego przy umiarkowanej insulinooporności, ale niższe wskaźniki były widoczne przy cięższej insulinooporności (Satapati i in., 4; Sunny i in., 2015). U otyłych ludzi ze stłuszczeniem wątroby szybkość ketogenezy jest normalna (Bickerton i in., 2012; Sunny i in., 2015), a zatem tempo ketogenezy jest zmniejszone w stosunku do zwiększonego obciążenia kwasami tłuszczowymi w hepatocytach. W konsekwencji acetylo-CoA pochodzący z a-oksydacji może być skierowany na końcowe utlenianie w cyklu TCA, zwiększając końcowe utlenianie, glukoneogenezę wywoływaną przez fosfoenolopirogronian poprzez anaplerozę / kataplerozę i stres oksydacyjny. Acetylo-CoA prawdopodobnie jest również eksportowany z mitochondriów jako cytrynian, prekursorowy substrat lipogenezy (ryc. 2012) (Satapati i in., 1; Satapati i in., 2016; Solinas i in., 1). Podczas gdy ketogeneza staje się mniej wrażliwa na insulinę lub post z przedłużającą się otyłością (Satapati i wsp., 2), podstawowe mechanizmy i dalsze konsekwencje tego pozostają nie w pełni zrozumiałe. Niedawne dowody wskazują, że mTORC2010 hamuje ketogenezę w sposób, który może następować za sygnalizacją insuliny (Kucejova i wsp., 2), co jest zgodne z obserwacjami, że mTORC1 hamuje indukcję HmgcsXNUMX pośredniczoną przez PPARy (Sengupta i wsp., XNUMX) ( zobacz także rozporządzenie HMGCSXNUMX i SCOT / OXCTXNUMX).
Wstępne obserwacje z naszej grupy sugerują niekorzystne następstwa niewydolności ketogennej dla wątroby (Cotter et al., 2014). Aby przetestować hipotezę, że upośledzona ketogeneza, nawet w stanach bogatych w węglowodany, a tym samym `` nieketogennych '', przyczynia się do nieprawidłowego metabolizmu glukozy i wywołuje stłuszczeniowe zapalenie wątroby, stworzyliśmy mysi model znacznej niewydolności ketogennej poprzez podanie antysensownych oligonukleotydów (ASO) ukierunkowanych na Hmgcs2. Utrata HMGCS2 u standardowych dorosłych myszy karmionych niskotłuszczową karmą spowodowała łagodną hiperglikemię i znacznie zwiększoną produkcję setek metabolitów wątrobowych, z których zestaw silnie sugerował aktywację lipogenezy. Karmienie myszy dietą wysokotłuszczową z niewystarczającą ketogenezą spowodowało rozległe uszkodzenie hepatocytów i stan zapalny. Odkrycia te potwierdzają główne hipotezy, że (i) ketogeneza nie jest pasywną ścieżką przepełnienia, ale raczej dynamicznym węzłem w wątrobowej i zintegrowanej fizjologicznej homeostazie, oraz (ii) rozważne wzmocnienie ketogeniczne w celu złagodzenia NAFLD / NASH i zaburzonego metabolizmu glukozy w wątrobie jest warte zbadania .
W jaki sposób zaburzona ketogeneza może przyczynić się do uszkodzenia wątroby i zmiany homeostazy glukozy? Pierwszą kwestią jest to, czy winowajcą jest niedobór strumienia ketogenicznego, czy też same ketony. Niedawny raport sugeruje, że ciała ketonowe mogą łagodzić uszkodzenie wątroby wywołane stresem oksydacyjnym w odpowiedzi na wielonienasycone kwasy tłuszczowe n-3 (Pawlak i in., 2015). Przypomnijmy, że ze względu na brak ekspresji SCOT w hepatocytach, ciała ketonowe nie są utleniane, ale mogą przyczyniać się do lipogenezy i pełnić różne funkcje sygnalizacyjne niezależne od ich utleniania (patrz także Nieoksydacyjne losy metaboliczne ciał ketonowych i OHB jako mediator sygnałowy). Możliwe jest również, że ciałka ketonowe pochodzące z hepatocytów mogą służyć jako sygnał i / lub metabolit dla sąsiednich typów komórek w obrębie wątroby trądzikowej, w tym komórek gwiaździstych i makrofagów komórek Kupffera. Chociaż dostępna ograniczona literatura sugeruje, że makrofagi nie są zdolne do utleniania ciał ketonowych, zostało to zmierzone tylko przy użyciu klasycznych metod i tylko w makrofagach otrzewnowych (Newsholme i in., 1986; Newsholme i in., 1987), co wskazuje, że re- ocena jest właściwa, biorąc pod uwagę obfitą ekspresję SCOT w makrofagach pochodzących ze szpiku kostnego (Youm et al., 2015).
Strumień ketogenny hepatocytów może również działać cytoprotekcyjnie. Chociaż zbawienne mechanizmy mogą nie zależeć od ketogenezy jako takiej, diety ketogenne o niskiej zawartości węglowodanów są związane z łagodzeniem NAFLD (Browning i wsp., 2011; Foster i wsp., 2010; Kani i wsp., 2014; Schugar i Crawford, 2012) . Nasze obserwacje wskazują, że ketogeneza hepatocytów może powodować sprzężenie zwrotne i regulować przepływ cyklu TCA, strumień anaplerotyczny, glukoneogenezę pochodzącą z fosfoenolopirogronianu (Cotter et al., 2014), a nawet przemianę glikogenu. Upośledzenie ketogeniczne kieruje acetylo-CoA w celu zwiększenia przepływu TCA, co w wątrobie jest powiązane ze zwiększonym uszkodzeniem, w którym pośredniczą ROS (Satapati i in., 2015; Satapati i in., 2012); wymusza przekierowanie węgla na zsyntetyzowane de novo gatunki lipidów, które mogą okazać się cytotoksyczne; i zapobiega ponownemu utlenianiu NADH do NAD + (Cotter i wsp., 2014) (Rys. 4). Podsumowując, przyszłe eksperymenty są potrzebne, aby zająć się mechanizmami, przez które względna niewydolność ketogeniczna może stać się nieprzystosowana, przyczynić się do hiperglikemii, wywołać stłuszczeniowe zapalenie wątroby oraz sprawdzić, czy mechanizmy te działają w ludzkim NAFLD / NASH. Ponieważ dowody epidemiologiczne wskazują na upośledzoną ketogenezę podczas progresji stłuszczeniowego zapalenia wątroby (Embade i wsp., 2016; Marinou i wsp., 2011; M nnist i wsp., 2015; Pramfalk i wsp., 2015; Safaei i wsp., 2016) terapie zwiększające ketogenezę wątrobową mogą okazać się zbawienne (Degirolamo i in., 2016; Honda i in., 2016).
Ciała ketonowe i niewydolność serca (HF)
Przy tempie metabolizmu przekraczającym 400 kcal/kg/dzień i obrocie 6 kg ATP/dzień serce jest organem o największym wydatku energetycznym i zapotrzebowaniu oksydacyjnym (Ashrafian i wsp., 35; Wang i wsp., 2007b). Zdecydowana większość obrotu energetycznego mięśnia sercowego znajduje się w mitochondriach, a 2010% tej podaży pochodzi z FAO. Serce jest wszystkożerne i elastyczne w normalnych warunkach, ale serce ulegające patologicznej przebudowie (np. z powodu nadciśnienia lub zawału mięśnia sercowego) i serce z cukrzycą stają się metabolicznie nieelastyczne (Balasse i Fery, 70; BING, 1989; Fukao i in., 1954 Lopaschuk i wsp., 2004; Taegtmeyer i wsp., 2010; Taegtmeyer i wsp., 1980; Young i wsp., 2002). Rzeczywiście, genetycznie zaprogramowane nieprawidłowości metabolizmu paliwa sercowego w modelach mysich wywołują kardiomiopatię (Carley i wsp., 2002; Neubauer, 2014). W warunkach fizjologicznych normalne serce utlenia ciała ketonowe proporcjonalnie do ich dostarczenia, kosztem utleniania kwasów tłuszczowych i glukozy, a mięsień sercowy jest największym konsumentem ciał ketonowych na jednostkę masy (BING, 2007; Crawford i in., 1954; GARLAND i in. 2009; Hasselbaink i wsp. 1962; Jeffrey i wsp. 2003; Pelletier i wsp. 1995; Tardif i wsp. 2007; Yan i wsp. 2001). W porównaniu z utlenianiem kwasów tłuszczowych, ciała ketonowe są bardziej wydajne energetycznie, dostarczając więcej energii dostępnej do syntezy ATP na cząsteczkę zainwestowanego tlenu (stosunek P/O) (Kashiwaya et al., 2009; Sato et al., 2010; Veech, 1995) . Utlenianie ciała ketonowego daje również potencjalnie wyższą energię niż FAO, utrzymując utlenianie ubichinonu, co zwiększa zakres redoks w łańcuchu transportu elektronów i udostępnia więcej energii do syntezy ATP (Sato i wsp., 2004; Veech, 1995). Utlenianie ciał ketonowych może również ograniczać produkcję ROS, a tym samym stres oksydacyjny (Veech, 2004).
Wstępne badania interwencyjne i obserwacyjne wskazują na potencjalną zbawienną rolę ciał ketonowych w sercu. W doświadczalnym kontekście uszkodzenia niedokrwienno-reperfuzyjnego ciałka ketonowe nadają potencjalne działanie kardioprotekcyjne (Al-Zaid i wsp., 2007, Wang i wsp., 2008), prawdopodobnie z powodu zwiększonej ilości mitochondrialnej w sercu lub regulacji w górę kluczowej fosforylacji oksydacyjnej mediatory (Snorek i wsp., 2012; Zou i wsp., 2002). Ostatnie badania wskazują, że wykorzystanie ciała ketonowego wzrasta w wadach serc myszy (Aubert i wsp., 2016) i ludzi (Bedi i wsp., 2016), wspierając wcześniejsze obserwacje u ludzi (BING, 1954, Fukao i wsp., 2000; Janardhan i wsp., 2011, Longo i wsp., 2004, Rudolph i Schinz, 1973, Tildon i Cornblath, 1972). Stężenia ketonów w cyrkulujących ciałach są zwiększone u pacjentów z niewydolnością serca, w bezpośredniej proporcji do ciśnień napełniania, obserwacje, których mechanizm i znaczenie pozostają nieznane (Kupari i wsp., 1995, Lommi i wsp., 1996, Lommi i wsp., 1997, Neely i in. 1972), ale myszy z selektywnym niedoborem SCOT w kardiomiocytach wykazują przyspieszoną patologiczną remodelację komorową i sygnatury ROS w odpowiedzi na chirurgicznie wywołane przeciążenie ciśnieniowe (Schugar i wsp., 2014).
Ostatnie intrygujące obserwacje w terapii cukrzycy ujawniły potencjalny związek między metabolizmem ketonowym mięśnia sercowego a patologiczną przebudową komory (Ryc. 5). Zahamowanie pobranego przez nerkowy proksymalny cylindryczny transporter sodu / glukozy 2 (SGLT2i) zwiększa stężenie w krążeniu ketonów u ludzi (Ferrannini i wsp., 2016a, Inagaki i wsp., 2015) i myszy (Suzuki i wsp., 2014) poprzez zwiększenie ketogeneza wątrobowa (Ferrannini i wsp., 2014, Ferrannini i wsp., 2016a, Katz and Leiter, 2015, Mudaliar i wsp., 2015). Co uderzające, co najmniej jeden z tych środków zmniejszył hospitalizację z powodu HF (np. Jak ujawniono w badaniu EMPA-REG OUTCOME) i zwiększoną śmiertelność z przyczyn sercowo-naczyniowych (Fitchett i wsp., 2016, Sonesson i wsp., 2016, Wu i wsp., 2016a ; Zinman i wsp., 2015). Podczas gdy mechanizmy napędzające korzystne wyniki HF w powiązanym SGLT2i pozostają aktywnie dyskutowane, korzyści z przeżycia są prawdopodobnie wieloczynnikowe, prospektywnie włączając ketozę, ale także zbawienny wpływ na masę ciała, ciśnienie krwi, poziom glukozy i kwasu moczowego, sztywność tętnic, współczulny układ nerwowy, osmotyczny diureza / zmniejszona objętość osocza i zwiększony hematokryt (Raz i Cahn, 2016, Vallon i Thomson, 2016). Podsumowując, przekonanie, że terapeutycznie zwiększający ketonemię u pacjentów z HF lub o wysokim ryzyku rozwoju HF, pozostaje kontrowersyjne, ale jest przedmiotem aktywnych badań w badaniach przedklinicznych i klinicznych (Ferrannini i wsp., 2016b, Kolwicz i wsp., 2016, Lopaschuk i Verma, 2016, Mudaliar i wsp., 2016, Taegtmeyer, 2016).
Ciała ketonowe w biologii raka
Związki między ciałami ketonowymi a rakiem szybko pojawiają się, ale badania zarówno na modelach zwierzęcych, jak i na ludziach przyniosły różne wnioski. Ponieważ metabolizm ketonów jest dynamiczny i reaguje na stan składników odżywczych, kuszące jest utrzymywanie biologicznych powiązań z rakiem, ze względu na potencjał precyzyjnych terapii żywieniowych. Komórki rakowe przechodzą metaboliczne przeprogramowanie w celu utrzymania szybkiej proliferacji i wzrostu komórek (DeNicola i Cantley, 2015, Pavlova i Thompson, 2016). Klasyczny efekt Warburga w metabolizmie komórek rakowych wynika z dominującej roli glikolizy i fermentacji kwasu mlekowego w celu transferu energii i kompensacji mniejszej zależności od oksydacyjnej fosforylacji i ograniczonego oddychania mitochondrialnego (De Feyter i wsp., 2016, Grabacka i wsp., 2016; Kang i wsp., 2015, Poff i wsp., 2014, Shukla i wsp., 2014). Węgiel glukozy jest kierowany głównie przez glikolizę, szlak pentozofosforanowy i lipogenezę, które razem zapewniają związki pośrednie niezbędne do ekspansji biomasy nowotworowej (Grabacka i wsp., 2016, Shukla i wsp., 2014, Yoshii i wsp., 2015). Adaptacja komórek nowotworowych do deprywacji glukozy odbywa się poprzez zdolność do wykorzystywania alternatywnych źródeł paliwa, w tym octanu, glutaminy i asparaginianu (Jaworski i wsp., 2016, Sullivan i wsp., 2015). Na przykład, ograniczony dostęp do pirogronianu ujawnia zdolność komórek rakowych do przekształcania glutaminy w acetylo-CoA przez karboksylację, zachowując zarówno potrzeby energetyczne, jak i anaboliczne (Yang i wsp., 2014). Ciekawą adaptacją komórek rakowych jest zastosowanie octanu jako paliwa (Comerford i wsp., 2014, Jaworski i wsp., 2016, Mashimo i wsp., 2014, Wright i Simone, 2016, Yoshii i wsp., 2015). Octan jest również substratem dla lipogenezy, która jest krytyczna dla proliferacji komórek nowotworowych, a wzrost tego lipogennego kanału jest związany z krótszym przeżyciem pacjenta i większym obciążeniem nowotworowym (Comerford i wsp., 2014, Mashimo i wsp., 2014; Yoshii i in. ., 2015).
Komórki nienowotworowe łatwo przenoszą swoje źródło energii z glukozy do ciał ketonowych podczas niedoboru glukozy. Ta plastyczność może być bardziej zmienna w różnych typach komórek rakowych, ale guzy mózgu wszczepione in vivo utleniają się [2,4-13C2] - aOHB w podobnym stopniu jak otaczająca tkanka mózgowa (De Feyter et al., 2016). Modele `` Odwrotnego efektu Warburga '' lub `` metabolizmu guza dwuprzedziałowego '' zakładają hipotezę, że komórki rakowe indukują produkcję OHB w sąsiednich fibroblastach, zaspokajając potrzeby energetyczne komórek nowotworowych (Bonuccelli et al., 2010; Martinez-Outschoorn et al., 2012) . W wątrobie, przesunięcie hepatocytów od ketogenezy do utleniania ketonów w komórkach raka wątrobowokomórkowego (wątrobiaka) jest zgodne z aktywacją aktywności BDH1 i SCOT obserwowaną w dwóch liniach komórkowych wątrobiaka (Zhang i wsp., 1989). Rzeczywiście, komórki wątrobiaka eksprymują OXCT1 i BDH1 i utleniają ketony, ale tylko wtedy, gdy brakuje surowicy (Huang i wsp., 2016). Alternatywnie zaproponowano również ketogenezę komórek nowotworowych. Dynamiczne zmiany w ekspresji genów ketogennych są widoczne podczas transformacji nowotworowej nabłonka okrężnicy, typu komórek, które normalnie eksprymują HMGCS2, a niedawny raport sugerował, że HMGCS2 może być markerem prognostycznym złego rokowania w raku jelita grubego i płaskonabłonkowego (Camarero i wsp., 2006; Chen i in., 2016). Pozostaje do ustalenia, czy ten związek wymaga ketogenezy, czy też funkcji księżycowej HMGCS2, czy też się z nią wiąże. I odwrotnie, pozorna produkcja αOHB przez komórki czerniaka i glejaka, stymulowana przez PPAR ™. agonista fenofibrat, był związany z zatrzymaniem wzrostu (Grabacka i wsp., 2016). Konieczne są dalsze badania, aby scharakteryzować role ekspresji HMGCS2 / SCOT, ketogenezy i utleniania ketonów w komórkach nowotworowych.
Poza dziedziną metabolizmu paliw, ketony zostały ostatnio zaangażowane w biologię komórek rakowych poprzez mechanizm sygnalizacyjny. Analiza czerniaka BRAF-V600E + wykazała zależną od OCT1 indukcję HMGCL w sposób onkogenny zależny od BRAF (Kang i wsp., 2015). Zwiększenie HMGCL było skorelowane z wyższym stężeniem AcAc w komórkach, co z kolei wzmocniło interakcję BRAFV600E-MEK1, wzmacniając sygnalizację MEK-ERK w pętli sprzężenia zwrotnego, która napędza proliferację i wzrost komórek nowotworowych. Obserwacje te nasuwają intrygujące pytanie o potencjalną pozawątrobową ketogenezę, która następnie wspiera mechanizm sygnalizacji (patrz także? OHB jako mediator sygnalizacji i kontrowersje w pozawątrobowej ketogenezie). Ważne jest również rozważenie niezależnego wpływu AcAc, d-aOHB i l-yOHB na metabolizm raka, a biorąc pod uwagę HMGCL, katabolizm leucyny może być również zaburzony.
Skutki diet ketogenicznych (patrz również Terapeutyczne zastosowanie diety ketogenicznej i egzogennych ciał ketonowych) w modelach zwierzęcych raka są zróżnicowane (De Feyter i in., 2016; Klement i in., 2016; Meidenbauer i in., 2015; Poff i in., 2014 ., 2011; Seyfried i wsp., 2014; Shukla i wsp., 2016). Podczas gdy powiązania epidemiologiczne między otyłością, rakiem i dietami ketogenicznymi są przedmiotem debaty (Liskiewicz i in., 2016; Wright i Simone, 2016), metaanaliza wykorzystująca diety ketogeniczne w modelach zwierzęcych i badaniach na ludziach sugeruje zbawienny wpływ na przeżycie, z korzyści prospektywnie związane z wielkością ketozy, czasem rozpoczęcia diety i lokalizacją guza (Klement i in., 2016; Woolf i in., 81). Leczenie komórek raka trzustki ciałami ketonowymi (d-aOHB lub AcAc) zahamowało wzrost, proliferację i glikolizę, a dieta ketogeniczna (18% kcal tłuszczu, 1% białka, 2014% węglowodanów) zmniejszyła in vivo masę guza, glikemię i wzrost masy mięśniowej i ciała u zwierząt z wszczepionym rakiem (Shukla i wsp., 2014). Podobne wyniki zaobserwowano przy użyciu modelu komórek przerzutowych glejaka u myszy, które otrzymały suplementację ketonową w diecie (Poff i wsp., 91). I odwrotnie, dieta ketogeniczna (9% kcal tłuszczu, 2016% białka) zwiększyła stężenie krążącego ?OHB i zmniejszyła glikemię, ale nie miała wpływu ani na objętość guza, ani na czas przeżycia u szczurów z glejakiem (De Feyter i wsp., 2015). Jako wskaźnik kliniczny, który poprawia zarządzanie metaboliczne w terapii raka mózgu wywołanego dietą ketogenną u ludzi i myszy, zaproponowano indeks glukozowo-ketonowy (Meidenbauer i in., XNUMX). Podsumowując, role metabolizmu ciał ketonowych i ciał ketonowych w biologii raka są kuszące, ponieważ każdy z nich stanowi wykonalne opcje terapeutyczne, ale podstawowe aspekty pozostają do wyjaśnienia, z wyraźnymi wpływami wyłaniającymi się z macierzy zmiennych, w tym (i) różnic między egzogennymi ketonami organizmy a dieta ketogeniczna, (ii) typ komórek rakowych, polimorfizmy genomowe, stopień i stadium zaawansowania; oraz (iii) czas i czas trwania ekspozycji na stan ketotyczny.
Ketogeneza jest tworzona przez ciała ketonowe poprzez rozkład kwasów tłuszczowych i aminokwasów ketogennych. Ten biochemiczny proces dostarcza energii do różnych narządów, szczególnie do mózgu, w warunkach postu jako odpowiedź na niedostępność glukozy we krwi. Ciała ketonowe są produkowane głównie w mitochondriach komórek wątroby. Podczas gdy inne komórki są zdolne do przeprowadzenia ketogenezy, nie są tak skuteczne w robieniu takich komórek jak komórki wątroby. Ponieważ ketogeneza występuje w mitochondriach, jej procesy są regulowane niezależnie. Dr Alex Jimenez DC, CCST Insight
Terapeutyczne zastosowanie diety ketogenicznej i egzogennych ciał ketonowych
Zastosowania diet ketogenicznych i ciał ketonowych jako narzędzi terapeutycznych pojawiły się również w kontekstach nienowotworowych, w tym otyłości i NAFLD/NASH (Browning i in., 2011; Foster i in., 2010; Schugar i Crawford, 2012); niewydolność serca (Huynh, 2016; Kolwicz i in., 2016; Taegtmeyer, 2016); choroby neurologiczne i neurodegeneracyjne (Martin i in., 2016; McNally i Hartman, 2012; Rho, 2015; Rogawski i in., 2016; Yang i Cheng, 2010; Yao i in., 2011); wrodzone błędy metabolizmu (Scholl-B�rgi i in., 2015); i wydajność ćwiczeń (Cox i in., 2016). Skuteczność diet ketogenicznych została szczególnie doceniona w terapii napadów padaczkowych, zwłaszcza u pacjentów lekoopornych. W większości badań oceniano diety ketogeniczne u pacjentów pediatrycznych i wykazano do ~50% zmniejszenie częstości napadów po 3 miesiącach, z lepszą skutecznością w wybranych zespołach (Wu i wsp., 2016b). Doświadczenie jest bardziej ograniczone w przypadku padaczki dorosłych, ale widoczna jest podobna redukcja, z lepszą odpowiedzią u pacjentów z objawową padaczką uogólnioną (Nei et al., 2014). Podstawowe mechanizmy przeciwdrgawkowe pozostają niejasne, chociaż postulowane hipotezy obejmują zmniejszone wykorzystanie glukozy/glikolizę, przeprogramowany transport glutaminianu, pośredni wpływ na wrażliwy na ATP kanał potasowy lub receptor adenozyny A1, zmianę ekspresji izoformy kanału sodowego lub wpływ na krążące hormony, w tym leptynę ( Lambrechts i wsp., 2016; Lin i wsp., 2017; Lutas i Yellen, 2013). Pozostaje niejasne, czy efekt przeciwdrgawkowy można przypisać przede wszystkim ciałkom ketonowym, czy też kaskadowym metabolicznym konsekwencjom diety niskowęglowodanowej. Niemniej jednak estry ketonowe (patrz poniżej) wydają się podnosić próg drgawkowy w zwierzęcych modelach napadów prowokowanych (Ciarlone i in., 2016; D'Agostino i in., 2013; Viggiano i in., 2015).
Dieta w stylu Atkinsa i ketogeniczna dieta o niskiej zawartości węglowodanów są często uważane za nieprzyjemne i mogą powodować zaparcia, hiperurykemię, hipokalcemię, hipomagnezemię, prowadzić do kamicy nerkowej, kwasicy ketonowej, powodować hiperglikemię oraz zwiększać stężenie cholesterolu we krwi i wolnego kwasu tłuszczowego (Bisschop i in., 2001 Kossoff i Hartman, 2012, Kwiterovich i wsp., 2003, Suzuki i wsp., 2002). Z tych powodów długotrwałe stosowanie stawia wyzwania. Badania gryzoni często wykorzystują charakterystyczny rozkład makroskładników (94% kcal, 1% kcal węglowodan, 5% kcal protein, Bio-Serv F3666), który wywołuje silną ketozę. Jednak zwiększenie zawartości białka nawet do 10% kcal znacznie zmniejsza ketozę, a ograniczenie białka 5% kcal wywołuje zakłócające efekty metaboliczne i fizjologiczne. Ten preparat diety jest także pozbawiony choliny, inna zmienna, która wpływa na podatność na uszkodzenie wątroby, a nawet ketogenezę (Garbow i wsp., 2011, Jornayvaz i wsp., 2010, Kennedy i wsp., 2007, Pissios i wsp., 2013; Schugar; i wsp., 2013). Skutki długotrwałego stosowania diet ketogennych u myszy nie zostały jeszcze w pełni określone, ale ostatnie badania na myszach wykazały normalne przeżycie i brak markerów uszkodzenia wątroby u myszy na dietach ketogennych w ciągu ich życia, chociaż metabolizm aminokwasów, wydatek energetyczny i sygnalizacja insulinowa zostały znacznie przeprogramowane (Douris et al., 2015).
Mechanizmy zwiększające ketozę poprzez mechanizmy alternatywne dla diet ketogennych obejmują stosowanie prekursorów ciała ketonowego do spożycia. Podawanie egzogennych ciał ketonowych może stworzyć wyjątkowy stan fizjologiczny, nie spotykany w normalnej fizjologii, ponieważ stężenia glukozy i insuliny w krążeniu są względnie normalne, podczas gdy komórki mogą oszczędzić wychwytu glukozy i jej wykorzystania. Same ciała ketonowe mają krótki okres półtrwania, a spożycie lub wlew soli sodowej a-OHB w celu uzyskania terapeutycznej ketozy wywołuje niepożądane obciążenie sodem. R / S-1,3-butanodiol jest nietoksycznym dialkoholem, który łatwo utlenia się w wątrobie z wytworzeniem d / l-aOHB (Desrochers i wsp., 1992). W różnych kontekstach eksperymentalnych tę dawkę podawano codziennie myszom lub szczurom nawet przez siedem tygodni, uzyskując krążące stężenie OHB do 5 mM w ciągu 2 godzin od podania, które jest stabilne przez co najmniej dodatkowe 3 godziny (D ' Agostino i in., 2013). Częściowe zahamowanie przyjmowania pokarmu obserwowano u gryzoni, którym podawano R / S-1,3-butanodiol (Carpenter and Grossman, 1983). Ponadto trzy chemicznie różne estry ketonowe (KE), (i) monoester R-1,3-butanodiolu i d-a-OHB (R-3-hydroksybutylo-R-a-OHB); (ii) glicerylo-tris-aOHB; oraz (iii) diester acetooctanowy R, S-1,3-butanodiolu, również był szeroko badany (Brunengraber, 1997; Clarke i in., 2012a; Clarke i in., 2012b; Desrochers i in., 1995a; Desrochers i in. ., 1995b; Kashiwaya i in., 2010). Nieodłączną zaletą tego pierwszego jest to, że po hydrolizie esterazy w jelicie lub wątrobie wytwarzane są 2 mole fizjologicznej d-aOHB na mol KE. Bezpieczeństwo, farmakokinetykę i tolerancję najszerzej badano u ludzi przyjmujących R-3-hydroksybutylo R-aOHB w dawkach do 714 mg / kg, uzyskując krążące stężenia d-a-OHB do 6 mM (Clarke et al., 2012a; Cox i in., 2016; Kemper i in., 2015; Shivva i in., 2016). U gryzoni ten KE zmniejsza kaloryczność i całkowity cholesterol w osoczu, stymuluje brązową tkankę tłuszczową i polepsza oporność na insulinę (Kashiwaya i wsp., 2010, Kemper i wsp., 2015, Veech, 2013). Niedawne odkrycia wskazują, że podczas ćwiczeń u wytrenowanych sportowców spożycie R-3-hydroksybutylo R-aOHB zmniejszyło glikolizę mięśni szkieletowych i stężenie mleczanu w osoczu, zwiększyło domięśniowe utlenianie triacyloglicerolu i zachowało zawartość glikogenu w mięśniach, nawet gdy jednocześnie spożywane węglowodany stymulowały wydzielanie insuliny ( Cox i in., 2016). Konieczny jest dalszy rozwój tych intrygujących wyników, ponieważ poprawa wydajności ćwiczeń wytrzymałościowych była głównie spowodowana silną odpowiedzią na KE u pacjentów 2 / 8. Niemniej jednak, wyniki te potwierdzają klasyczne badania wskazujące na preferencję do utleniania ketonów względem innych substratów (GARLAND i wsp., 1962, Hasselbaink i wsp., 2003, Stanley i wsp., 2003, Valente-Silva i wsp., 2015), w tym podczas ćwiczeń, i że wyszkoleni sportowcy mogą być bardziej przygotowani do wykorzystania ketonów (Johnson i wsp., 1969a, Johnson i Walton, 1972, Winder i wsp., 1974, Winder i wsp., 1975). Wreszcie, należy określić mechanizmy, które mogą wspierać poprawę wydajności wysiłkowej po równym spożyciu kalorii (zróżnicowane rozmieszczenie wśród makroelementów) i równe wskaźniki zużycia tlenu.
Przyszłe perspektywy
Niedawno napiętnowane jako ścieżka przelewowa zdolna do akumulacji toksycznych emisji ze spalania tłuszczu w stanach ograniczenia węglowodanów (paradygmat „ketotoksyczny”), ostatnie obserwacje potwierdzają pogląd, że metabolizm ciała ketonowego pełni zbawienną rolę nawet w stanach obciążonych węglowodanami, otwierając „ketohormetykę”. hipoteza. Podczas gdy łatwe podejście żywieniowe i farmakologiczne do manipulowania metabolizmem ketonów czyni go atrakcyjnym celem terapeutycznym, agresywnie nastawione, ale rozważne eksperymenty pozostają w laboratoriach badawczych zarówno podstawowych, jak i translacyjnych. Niezaspokojone potrzeby pojawiły się w dziedzinach definiowania roli dźwigni metabolizmu ketonów w niewydolności serca, otyłości, NAFLD/NASH, cukrzycy typu 2 i raku. Zakres i wpływ „niekanonicznych� ról sygnalizacyjnych ciał ketonowych, w tym regulacja PTM, które prawdopodobnie sprzężą się w przód i w tył na szlaki metaboliczne i sygnalizacyjne, wymagają głębszej eksploracji. Wreszcie, ketogeneza pozawątrobowa może otworzyć intrygujące parakrynne i autokrynne mechanizmy sygnalizacyjne oraz możliwości wpływania na kometabolizm w obrębie układu nerwowego i guzów w celu osiągnięcia celów terapeutycznych.
Podsumowując, ciała ketonowe są tworzone przez wątrobę, aby mogły być wykorzystywane jako źródło energii, gdy w organizmie ludzkim nie ma wystarczającej ilości łatwo dostępnej glukozy. Ketogeneza zachodzi, gdy we krwi występuje niski poziom glukozy, szczególnie po wyczerpaniu innych komórkowych zapasów węglowodanów. Celem powyższego artykułu było omówienie wielowymiarowej roli ciał ketonowych w metabolizmie paliw, sygnalizacji i terapii. Zakres naszych informacji ogranicza się do zagadnień związanych ze zdrowiem kręgosłupa i kręgosłupa. Aby omówić ten temat, prosimy zapytać dr Jimeneza lub skontaktować się z nami pod adresem 915-850-0900 .
Na ból plecówTo jedna z najczęstszych przyczyn niepełnosprawności i nieobecności w pracy na całym świecie. Ból pleców jest drugą najczęstszą przyczyną wizyt lekarskich, ustępującą jedynie infekcjom górnych dróg oddechowych. Około 80 procent populacji przynajmniej raz w życiu doświadczy bólu pleców. Kręgosłup to złożona struktura składająca się z kości, stawów, więzadeł i mięśni oraz innych tkanek miękkich. Urazy i/lub pogorszenie stanu, takie jak�przepukliny, może ostatecznie prowadzić do objawów bólu pleców. Urazy sportowe lub urazy powypadkowe są często najczęstszą przyczyną bólu pleców, jednak czasami najprostsze ruchy mogą mieć bolesne skutki. Na szczęście alternatywne opcje leczenia, takie jak chiropraktyka, mogą pomóc złagodzić ból pleców poprzez zastosowanie korekty kręgosłupa i manipulacji manualnych, ostatecznie poprawiając ulgę w bólu. �
Połączenia szlak sygnałowy czynnika 2 związany z jądrowym erytroidem 2, najlepiej znany jako Nrf2, jest mechanizmem ochronnym, który działa jako „główny regulator” odpowiedzi antyoksydacyjnej ludzkiego organizmu. Nrf2 wykrywa poziom stresu oksydacyjnego w komórkach i uruchamia ochronne mechanizmy antyoksydacyjne. Podczas gdy aktywacja Nrf2 może przynieść wiele korzyści, „nadekspresja” Nrf2 może wiązać się z kilkoma zagrożeniami.
Wydaje się, że zrównoważony poziom NRF2 ma zasadnicze znaczenie dla zapobiegania ogólnemu rozwojowi różnych chorób, oprócz ogólnej poprawy tych problemów zdrowotnych. Jednak NRF2 może również powodować komplikacje. Główną przyczyną „nadekspresji” NRF2 jest między innymi mutacja genetyczna lub ciągła przewlekła ekspozycja na stres chemiczny lub oksydacyjny. Poniżej omówimy wady nadekspresji Nrf2 i zademonstrujemy jej mechanizmy działania w organizmie człowieka.
Rak
Badania naukowe wykazały, że myszy, które nie wykazują ekspresji NRF2 są bardziej skłonne do rozwoju raka w odpowiedzi na stymulację fizyczną i chemiczną. Podobne badania wykazały jednak, że nadmierna aktywacja NRF2, a nawet inaktywacja KEAP1 może powodować zaostrzenie niektórych nowotworów, szczególnie jeśli te ścieżki zostały przerwane. Nadaktywność NRF2 może wystąpić podczas palenia, gdzie uważa się, że ciągła aktywacja NRF2 jest przyczyną raka płuc u palaczy. Nadekspresja Nrf2 może spowodować, że komórki rakowe nie ulegną samozniszczeniu, podczas gdy okresowa aktywacja NRF2 może zapobiec indukcji toksyn przez komórki rakowe.
Dodatkowo, ponieważ nadekspresja NRF2 zwiększa zdolność antyoksydacyjną organizmu człowieka do funkcjonowania poza homeostazą redoks, zwiększa to podział komórek i generuje nienaturalny wzór DNA i metylacji histonów. Może to ostatecznie sprawić, że chemioterapia i radioterapia będą mniej skuteczne w leczeniu raka. Dlatego ograniczenie aktywacji NRF2 za pomocą substancji takich jak DIM, Luteolin, Zi Cao lub salinomycyna może być idealne dla pacjentów z rakiem, chociaż nadaktywacja Nrf2 nie powinna być uważana za jedyną przyczynę raka. Niedobory składników odżywczych mogą wpływać na geny, w tym NRF2. Może to być jeden ze sposobów, w jaki niedobory przyczyniają się do powstawania guzów.
Wątroba
Nadmierna aktywacja Nrf2 może również wpływać na funkcję określonych narządów w ludzkim ciele. Nadekspresja NRF2 może ostatecznie zablokować produkcję insulinopodobnego czynnika wzrostu 1, czyli IGF-1, z wątroby, który jest niezbędny do regeneracji wątroby.
Serce
Podczas gdy ostra nadekspresja Nrf2 może przynosić korzyści, ciągła nadekspresja NRF2 może powodować długotrwały szkodliwy wpływ na serce, taki jak kardiomiopatia. Ekspresję NRF2 można zwiększyć poprzez wysoki poziom cholesterolu lub aktywację HO-1. Uważa się, że jest to powód, dla którego przewlekle podwyższony poziom cholesterolu może powodować problemy ze zdrowiem sercowo-naczyniowym.
bielactwo
Wykazano również, że nadekspresja NRF2 hamuje zdolność do repigmentacji w bielactwie, ponieważ może blokować działanie tyrozynazy lub TYR, które jest niezbędne do repigmentacji poprzez melaninogenezę. Badania naukowe wykazały, że proces ten może być jednym z głównych powodów, dla których osoby z bielactwem nie wydają się aktywować Nrf2 tak skutecznie, jak osoby bez bielactwa.
Dlaczego NRF2 może nie działać prawidłowo
Hormesis
NRF2 musi być aktywowany hormetycznie, aby móc skorzystać z jego zalet. Innymi słowy, Nrf2 nie powinien wyzwalać się co minutę ani codziennie, dlatego warto robić sobie przerwy, na przykład 5 dni w 5 dni wolne lub co drugi dzień. NRF2 musi również osiągnąć określony próg, aby wywołać reakcję hormonalną, w której mały stresor może nie wystarczyć, aby ją wywołać.
DJ-1 Utlenianie
Deglikaza białkowa DJ-1 lub po prostu DJ-1, zwana także białkiem choroby Parkinsona lub PARK7, jest głównym regulatorem i detektorem stanu redoks w ludzkim ciele. DJ-1 jest niezbędny do regulowania, jak długo NRF2 może pełnić swoją funkcję i wywoływać odpowiedź antyoksydacyjną. W przypadku, gdy DJ-1 zostanie nadtleniony, komórki sprawią, że białko DJ-1 będzie mniej dostępne.
Proces ten powoduje, że aktywacja NRF2 wygasa zbyt szybko, ponieważ DJ-1 ma kluczowe znaczenie dla utrzymania zrównoważonych poziomów NRF2 i zapobiegania ich rozkładowi w komórce. W przypadku, gdy białko DJ-1 nie istnieje lub jest nadtlenione, ekspresja NRF2 będzie prawdopodobnie minimalna, nawet przy użyciu DIM lub alternatywnych aktywatorów NRF2. Ekspresja DJ-1 jest niezbędna do przywrócenia zaburzonego działania NRF2.
Przewlekłe choroby
Jeśli masz przewlekłą chorobę, w tym CIRS, przewlekłe infekcje/dysbiozę/SIBO lub nagromadzenie metali ciężkich, takich jak rtęć i/lub z kanałów korzeniowych, mogą one blokować system NRF2 i fazę drugiej detoksykacji. Zamiast stresu oksydacyjnego przekształcającego NRF2 w przeciwutleniacz, NRF2 nie wyzwala, a stres oksydacyjny może pozostać w komórce i powodować uszkodzenia, co oznacza, że nie ma odpowiedzi antyoksydacyjnej. Jest to istotny powód, dla którego wiele osób z CIRS ma kilka wrażliwości i sięga do wielu czynników. Niektórzy uważają, że mogą mieć odpowiedź herx, jednak ta reakcja może tylko dalej uszkadzać komórki.
Jednak leczenie przewlekłej choroby pozwoli wątrobie na odprowadzanie toksyn do żółci, stopniowo rozwijając hormetyczną odpowiedź aktywacji NRF2. Jeśli żółć pozostaje toksyczna i nie jest wydalana z organizmu człowieka, reaktywuje stres oksydacyjny NRF2 i powoduje pogorszenie samopoczucia po ponownym wchłonięciu z przewodu pokarmowego. Na przykład ochratoksyna A może blokować NRF2. Oprócz leczenia problemu, inhibitory deacetylazy histonowej mogą blokować reakcję oksydacyjną z wielu czynników, które wyzwalają aktywację NRF2, ale mogą również zapobiegać normalnemu wyzwalaniu NRF2, co może ostatecznie nie spełniać swojego celu.
Rozregulowanie oleju rybiego
Cholinergiki to substancje, które pobudzają acetylocholinę lub ACh i cholinę w mózgu poprzez wzrost ACh, szczególnie podczas hamowania rozpadu ACh. Pacjenci z CIRS często mają problemy z rozregulowaniem poziomu acetylocholiny w organizmie człowieka, zwłaszcza w mózgu. Olej rybi wyzwala NRF2, aktywując jego ochronny mechanizm antyoksydacyjny w komórkach.
Osoby z chorobami przewlekłymi mogą mieć problemy ze stresem poznawczym i ekscytotoksycznością acetylocholiny, wynikającą z akumulacji organofosforanów, które mogą powodować stan zapalny w ludzkim ciele tranu. Niedobór choliny dodatkowo indukuje aktywację NRF2. Włączenie choliny do diety (polifenoli, jajek itp.) może pomóc wzmocnić efekty dysregulacji cholinergicznej.
Co zmniejsza NRF2?
Zmniejszenie nadekspresji NRF2 jest najlepsze dla osób z rakiem, chociaż może być korzystne w przypadku wielu innych problemów zdrowotnych.
Dieta, suplementy i popularne leki:
Apigenina (wyższe dawki)
Brucea Javanica
Kasztany
EGCG (wysokie dawki zwiększają NRF2)
Kozieradka (trygonellina)
Hiba (Hinokitiol / ?-tujaplicyna)
Dieta bogata w sól
Luteolina (seler, zielona papryka, pietruszka, liść perilli i herbata rumiankowa – wyższe dawki mogą zwiększyć NRF2 – 40 mg/kg luteoliny trzy razy w tygodniu)
Metformina (przewlekłe przyjmowanie)
N-Acetylo-L-Cysteina (NAC, poprzez blokowanie odpowiedzi oksydacyjnej, szczególnie przy wysokich dawkach)
Kwercetyna (większe dawki mogą zwiększyć NRF2 – 50 mg/kg/d kwercetyny)
Salinomycyna (lek)
Retinol (kwas all-trans retinowy)
Witamina C w połączeniu z kwercetyną
Zi Cao (Fioletowy Gromwel ma Shikonin/Alkannin)
Ścieżki i inne:
Bacha1
ZAKŁAD
Biofilmy
Brusatol
Kamptotecyna
DNMT
DPP-23
EZH2
Sygnalizacja receptora glukokortykoidowego (również deksametazon i betametazon)
GSK-3? (informacje regulacyjne)
Aktywacja HDAC?
Halofuginon
Homocysteina (ALCAR może odwrócić tę homocysteinę, indukując niski poziom NRF2)
IL-24
Zachowaj1
MDA-7
NF?B?
Ochratoksyna A (gatunki aspergillus i pencicllium)
Białko białaczki promielocytowej
p38
p53
p97
Receptor kwasu retinowego alfa
Selenit
SYVN1 (Hrd1)
Hamowanie STAT3 (takie jak kryptotanszynon)
Testosteron (i propionian testosteronu, chociaż TP donosowo może zwiększać NRF2)
Trecator (Etionamid)
Trx1 (poprzez redukcję Cys151 w Keap1 lub Cys506 w regionie NLS Nrf2)
Trolox
Vorinostat
Niedobór cynku (pogarsza go w mózgu)
Mechanizm działania Nrf2
Stres oksydacyjny wyzwala się poprzez CUL3, gdzie NRF2 z KEAP1, inhibitora ujemnego, następnie wnika do jądra tych komórek, stymulując transkrypcję ARE, zamieniając siarczki w dwusiarczki i zamieniając je w bardziej geny przeciwutleniające, co prowadzi do zwiększenia aktywności przeciwutleniaczy, takich jak jako GSH, GPX, GST, SOD itp. Resztę można zobaczyć na poniższej liście:
Zwiększa AKR
Zwiększa ARE
Zwiększa ATF4
Zwiększa Bcl-xL
Zwiększa Bcl-2
Zwiększa BDNF
Zwiększa BRCA1
Zwiększa c-cze
Zwiększa CAT
Zwiększa cGMP
Zwiększa CKIP-1
Zwiększa CYP450
Zwiększa Cul3
Zwiększa GCL
Zwiększa GCLC
Zwiększa GCLM
Zwiększa GCS
Zwiększa GPx
Zwiększa GR
Zwiększa GSH
Zwiększa GST
Zwiększa HIF1
Zwiększa HO-1
Zwiększa HQO1
Zwiększa HSP70
Zwiększa IL-4
Zwiększa IL-5
Zwiększa IL-10
Zwiększa IL-13
Zwiększa K6
Zwiększa K16
Zwiększa K17
Zwiększa mEH
Zwiększa Mrp2-5
Zwiększa NADPH
Zwiększa wycięcie 1
Zwiększa NQO1
Zwiększa PPAR-alfa
Zwiększa Prx
Zwiększa p62
Zwiększa Sesn2
Zwiększa Slco1b2
Zwiększa sMafs
Zwiększa SOD
Zwiększa Trx
Zwiększa Txn(d)
Zwiększa UGT1 (A1/6)
Zwiększa VEGF
Zmniejsza ADAMTS(4/5)
Zmniejsza alfa-SMA
Zmniejsza ALT
Zmniejsza AP1
Zmniejsza AST
Redukuje Bacha1
Zmniejsza COX-2
Zmniejsza DNMT
Zmniejsza FASN
Zmniejsza FGF
Zmniejsza HDAC
Zmniejsza IFN-?
Zmniejsza IgE
Zmniejsza IGF-1
Zmniejsza IL-1b
Redukuje IL-2
Redukuje IL-6
Redukuje IL-8
Redukuje IL-25
Redukuje IL-33
Zmniejsza iNOS
Zmniejsza LT
Zmniejsza utrzymanie1
Zmniejsza MCP-1
Zmniejsza MIP-2
Zmniejsza MMP-1
Zmniejsza MMP-2
Zmniejsza MMP-3
Zmniejsza MMP-9
Zmniejsza MMP-13
Zmniejsza NfkB
Redukuje NO
Zmniejsza SIRT1
Zmniejsza TGF-b1
Zmniejsza TNF-alfa
Zmniejsza Tyr
Zmniejsza VCAM-1
Zakodowany z genu NFE2L2, NRF2 lub czynnika 2 związanego z jądrowym erytroidem 2, jest czynnikiem transkrypcyjnym w nadrodzinie podstawowego suwaka leucynowego lub bZIP, który wykorzystuje strukturę Cap'n'Collar lub CNC.
Promuje enzymy azotowe, enzymy biotransformacji i ksenobiotyczne transportery wypływowe.
Jest niezbędnym regulatorem w indukcji genów enzymów antyoksydacyjnych i detoksykacyjnych fazy II, które chronią komórki przed uszkodzeniami spowodowanymi stresem oksydacyjnym i atakami elektrofilowymi.
W warunkach homeostatycznych, Nrf2 jest sekwestrowany w cytozolu poprzez cielesne przyłączenie domeny N-końcowej Nrf2 lub białka związanego z ECH typu Kelch lub Keap1, zwanego również INrf2 lub Inhibitorem Nrf2, hamując aktywację Nrf2.
Może być również kontrolowany przez ssaczą selenoproteinową reduktazę tioredoksynową 1 lub TrxR1, która działa jako regulator ujemny.
Podatny na stresory elektrofilowe, Nrf2 dysocjuje od Keap1, przemieszczając się do jądra, gdzie następnie heterodimeryzuje z szeregiem białek regulatorowych transkrypcji.
Częste interakcje obejmują te z władzami transkrypcyjnymi Jun i Fos, które mogą być członkami rodziny białek aktywujących czynniki transkrypcyjne.
Po dimeryzacji kompleksy te wiążą się następnie ze składnikami reagującymi na przeciwutleniacze/elektrofile ARE/EpRE i aktywują transkrypcję, tak jak w przypadku kompleksu Jun-Nrf2 lub tłumią transkrypcję, podobnie jak kompleks Fos-Nrf2.
Pozycjonowanie ARE, które jest wyzwalane lub hamowane, określi, które geny są kontrolowane przez te zmienne transkrypcyjnie.
Po uruchomieniu ARE:
Aktywacja syntezy antyoksydantów jest zdolna do detoksykacji ROS, takich jak katalaza, dysmutaza ponadtlenkowa lub SOD, peroksydazy GSH, reduktaza GSH, transferaza GSH, oksydoreduktaza NADPH-chinonowa lub NQO1, system monooksygenazy cytochromu P450, tioredoksyna, tioredoksyna reduktaza i HSP70.
Aktywacja tej syntazy GSH umożliwia zauważalny wzrost wewnątrzkomórkowego stopnia GSH, co jest dość ochronne.
Zwiększenie tej syntezy i stopni enzymów fazy II, takich jak UDP-glukuronozylotransferaza, N-acetylotransferazy i sulfotransferazy.
Regulacja w górę HO-1, który jest naprawdę ochronnym receptorem z potencjalnym wzrostem CO, który w połączeniu z NO umożliwia rozszerzenie naczyń krwionośnych niedokrwionych komórek.
Redukcja przeładowania żelazem poprzez podwyższony poziom ferrytyny i bilirubiny jako lipofilowego przeciwutleniacza. Oba białka fazy II wraz z przeciwutleniaczami są w stanie naprawić przewlekły stres oksydacyjny, a także przywrócić normalny system redoks.
GSK3? pod kierownictwem AKT i PI3K fosforyluje Fyn, co skutkuje lokalizacją jądrową Fyn, która to fosforyluje Nrf2Y568, prowadząc do eksportu jądrowego i degradacji Nrf2.
NRF2 tłumi również odpowiedź TH1/TH17 i wzbogaca odpowiedź TH2.
Inhibitory HDAC wyzwalały ścieżkę sygnalizacyjną Nrf2 i regulowały w górę, że poniżej Nrf2 celuje w HO-1, NQO1 i podjednostkę katalityczną ligazy glutaminiano-cysteinowej lub GCLC, poprzez hamowanie Keap1 i zachęcanie do dysocjacji Keap1 od nuklearnej translokacji Nrf2, Nrf2 i Nrf2 -SĄ wiążące.
Nrf2 obejmuje okres półtrwania około 20 minut w warunkach podstawowych.
Zmniejszenie IKK? pula poprzez wiązanie Keap1 zmniejsza I?B? degradacji i może być nieuchwytnym mechanizmem, za pomocą którego, jak udowodniono, aktywacja Nrf2 hamuje aktywację NFkB.
Keap1 nie zawsze musi być regulowany w dół, aby NRF2 mógł działać, takie jak chlorofilina, jagoda, kwas elagowy, astaksantyna i polifenole herbaty mogą zwiększyć NRF2 i KEAP1 o 400 procent.
Nrf2 reguluje ujemnie poprzez termin desaturazy stearoilo-CoA (SCD) i liazy cytrynianowej (CL).
Genetyka
ZACHOWAJ1
rs1048290
allel C – wykazał znaczne ryzyko i działanie ochronne przeciwko padaczce lekoopornej (DRE)
rs11085735 (jestem AC)
związane z szybkością pogarszania się czynności płuc w LHS
MAPATY
rs242561
Allel T – allel ochronny dla chorób Parkinsona – miał silniejsze wiązanie NRF2/sMAF i był związany z wyższymi poziomami mRNA MAPT w 3 różnych obszarach mózgu, w tym w korze móżdżku (CRBL), korze skroniowej (TCTX), wewnątrzzrazikowej istocie białej (WHMT)
NFE2L2 (NRF2)
rs10183914 (jestem CT)
Allel T – podwyższony poziom białka Nrf2 i opóźniony wiek zachorowania na chorobę Parkinsona o cztery lata
rs16865105 (jestem AC)
Allel C – miał wyższe ryzyko choroby Parkinsona
rs1806649 (jestem CT)
Allel C – został zidentyfikowany i może mieć znaczenie dla etiologii raka piersi.
związane ze zwiększonym ryzykiem hospitalizacji w okresach wysokiego stężenia PM10
rs1962142 (jestem GG)
Allel T – wiązał się z niskim poziomem ekspresji cytoplazmatycznej NRF2 (P = 0.036) i ujemną ekspresją sulfidoksyny (P = 0.042)
Allel – chroniony przed spadkiem przepływu krwi w przedramieniu (FEV) (wymuszona objętość wydechowa w ciągu jednej sekundy) w stosunku do palenia papierosów (p = 0.004)
rs2001350 (jestem TT)
Allel T – chroniony przed spadkiem FEV (natężona objętość wydechowa w ciągu jednej sekundy) w zależności od palenia papierosów (p = 0.004)
rs2364722 (jestem AA)
Allel – chroniony przed spadkiem FEV (natężona objętość wydechowa w ciągu jednej sekundy) w zależności od palenia papierosów (p = 0.004)
rs2364723
Allel C – związany ze znacznie obniżonym FEV u japońskich palaczy z rakiem płuc
rs2706110
Allel G – wykazał znaczne ryzyko i działanie ochronne przed padaczką lekooporną (DRE)
Allele AA – wykazywały istotnie obniżoną ekspresję KEAP1
allele AA – wiązały się ze zwiększonym ryzykiem raka piersi (p = 0.011)
rs2886161 (jestem TT)
Allel T – związany z chorobą Parkinsona
rs2886162
Allel – wiązał się z niską ekspresją NRF2 (p = 0.011; OR 1.988; CI 1.162), a genotyp AA wiązał się z gorszym przeżyciem (p = 3.400; HR 0.032; CI 1.687–1.047)
rs35652124 (jestem TT)
Allel – związany z wyższym związanym z wiekiem w momencie wystąpienia choroby Parkinsona w porównaniu z allelem G
allel C – miał podwyższony poziom białka NRF2
Allel T – miał mniej białka NRF2 i większe ryzyko chorób serca i ciśnienia krwi
rs6706649 (jestem CC)
Allel C – miał niższe białko NRF2 i zwiększał ryzyko choroby Parkinsona
rs6721961 (jestem GG)
Allel T – miał niższe białko NRF2
Allele TT – związek między paleniem papierosów u nałogowych palaczy a spadkiem jakości nasienia
allel TT – wiązał się ze zwiększonym ryzykiem raka piersi [P = 0.008; LUB, 4.656; przedział ufności (CI), 1.350] i allel T były związane z niskim stopniem ekspresji białka NRF16.063 (P = 2; OR 0.0003; CI 2.420–1.491) i ujemną ekspresją SRXN3.926 (P = 1; OR, 0.047; CI = 1.867–1.002)
Allel T – allel był również nominalnie związany z 28-dniową śmiertelnością związaną z ALI po zespole ogólnoustrojowej odpowiedzi zapalnej
Allel T – chroniony przed spadkiem FEV (natężona objętość wydechowa w ciągu jednej sekundy) w zależności od palenia papierosów (p = 0.004)
Allel G – związany ze zwiększonym ryzykiem ALI po poważnym urazie u Europejczyków i Afroamerykanów (iloraz szans, OR 6.44; 95% przedział ufności
Allele AA – związane z astmą infekcyjną
allele AA – wykazywały znacznie obniżoną ekspresję genu NRF2 i w konsekwencji zwiększone ryzyko raka płuc, zwłaszcza u osób, które kiedykolwiek paliły papierosy
allele AA – miały istotnie wyższe ryzyko rozwoju T2DM (OR 1.77; 95% CI 1.26, 2.49; p = 0.011) w porównaniu z osobami z genotypem CC
Allele AA – silny związek między gojeniem się ran a późną toksycznością promieniowania (związany ze znacznie wyższym ryzykiem wystąpienia późnych efektów u Afroamerykanów z tendencją do rasy kaukaskiej)
związane z doustną terapią estrogenową i ryzykiem żylnej choroby zakrzepowo-zatorowej u kobiet po menopauzie
rs6726395 (jestem AG)
Allel – chroniony przed spadkiem FEV1 (wymuszona objętość wydechowa w ciągu jednej sekundy) w zależności od statusu palenia papierosów (p = 0.004)
Allel – związany ze znacznie zmniejszonym FEV1 u japońskich palaczy z rakiem płuc
Allele GG – miały wyższy poziom NRF2 i zmniejszone ryzyko zwyrodnienia plamki żółtej
Allele GG – miały wyższą przeżywalność z Cholangiocarcinoma
rs7557529 (jestem CT)
Allel C – związany z chorobą Parkinsona
Stres oksydacyjny i inne stresory mogą powodować uszkodzenia komórek, które ostatecznie mogą prowadzić do różnych problemów zdrowotnych. Badania naukowe wykazały, że aktywacja Nrf2 może promować ochronny mechanizm antyoksydacyjny organizmu człowieka, jednak naukowcy dyskutowali, że nadekspresja Nrf2 może mieć ogromne ryzyko dla ogólnego stanu zdrowia i dobrego samopoczucia. Z nadaktywacją Nrf2 mogą również wystąpić różne rodzaje raka.
Dr Alex Jimenez DC, CCST Insight
Sulforafan i jego wpływ na raka, śmiertelność, starzenie się, mózg i zachowanie, choroby serca i nie tylko
Izotiocyjaniany to jedne z najważniejszych związków roślinnych, które możesz znaleźć w swojej diecie. W tym filmie przedstawiam dla nich najbardziej wyczerpujący argument, jaki kiedykolwiek powstał. Krótki czas koncentracji? Przejdź do swojego ulubionego tematu, klikając jeden z poniższych punktów czasowych. Pełna oś czasu poniżej.
Kluczowe sekcje:
00: 01: 14 - Rak i śmiertelność
00: 19: 04 - Starzenie
00: 26: 30 - Mózg i zachowanie
00: 38: 06 - Ostatnie podsumowanie
00: 40: 27 - Dawka
Pełna oś czasu:
00: 00: 34 - Wprowadzenie sulforaphane, główny punkt widzenia wideo.
00: 01: 14 - spożycie warzyw krzyżowych i redukcja śmiertelności z wszystkich przyczyn.
00: 02: 12 - ryzyko raka prostaty.
00: 02: 23 - ryzyko raka pęcherza moczowego.
00: 02: 34 - Rak płuc w ryzyku palaczy.
00: 02: 48 - ryzyko raka piersi.
00: 03: 13 - Hipotetyczny: co, jeśli masz już raka? (interwencyjne)
00:03:35 – Prawdopodobny mechanizm napędzający dane asocjacyjne dotyczące raka i śmiertelności.
00: 04: 38 - Sulforafan i rak.
00:05:32 – Dowody na zwierzętach wykazujące silny wpływ ekstraktu z kiełków brokułów na rozwój guza pęcherza moczowego u szczurów.
00: 06: 06 - Wpływ bezpośredniej suplementacji sulforafanu u pacjentów z rakiem prostaty.
00: 11: 20 - napój z kiełków brokułów zwiększa wydalanie benzenu o 61%.
00: 13: 31 - homogenat z kiełków brokułów zwiększa enzymy antyoksydacyjne w górnych drogach oddechowych.
00: 15: 45 - śmiertelność wśród roślin krzyżowych i śmiertelność z powodu chorób serca.
00: 16: 55 - proszek z kiełków brokułów poprawia poziom lipidów we krwi i ogólne ryzyko chorób serca u chorych na cukrzycę typu 2.
00:19:04 – Początek sekcji starzenia.
00:19:21 – Dieta wzbogacona w sulforafan wydłuża życie chrząszczy od 15 do 30% (w określonych warunkach).
00: 20: 34 - Znaczenie niskiego stanu zapalnego dla długowieczności.
00: 22: 05 - warzywa kapustne i kiełki w proszku z brokułów wydają się zmniejszać szeroki zakres markerów stanu zapalnego u ludzi.
00: 23: 40 - Podsumowanie filmu wideo: rak, sekcje starzenia
00: 24: 14 - Badania na myszach sugerują, że sulforafan może poprawić zdolność adaptacyjną układu odpornościowego w starszym wieku.
00:25:18 – Sulforafan poprawił wzrost włosów w mysim modelu łysienia. Zdjęcie o 00:26:10.
00: 26: 30 - Początek sekcji mózgu i zachowania.
00: 27: 18 - Wpływ wyciągu z kiełków brokułów na autyzm.
00: 27: 48 - Wpływ glukorafaniny na schizofrenię.
00: 28: 17 - Rozpoczęcie dyskusji o depresji (prawdopodobny mechanizm i badania).
00:31:21 – Badanie na myszach przy użyciu 10 różnych modeli depresji wywołanej stresem wykazało, że sulforafan jest podobnie skuteczny jak fluoksetyna (prozac).
00: 32: 00 - Badanie pokazuje, że bezpośrednie spożycie glukorafaniny u myszy jest podobnie skuteczne w zapobieganiu depresji w modelu stresu społecznego.
00: 33: 01 - Początek sekcji zwanej neurodegeneracją.
00: 35: 01 - Sulforafan wstrzyknięty natychmiast po TBI poprawia pamięć (badanie myszy).
00: 35: 55 - Sulforafan i plastyczność neuronalna.
00:36:32 – Sulforafan poprawia uczenie się w modelu cukrzycy typu II u myszy.
00:37:19 – Dystrofia mięśniowa Sulforaphane i Duchenne'a.
00: 37: 44 - Hamowanie miostatyny w komórkach satelitarnych mięśni (in vitro).
00: 38: 06 - późne nagranie wideo: śmiertelność i rak, uszkodzenie DNA, stres oksydacyjny i zapalenie, wydalanie benzenu, choroba sercowo-naczyniowa, cukrzyca typu II, wpływ na mózg (depresja, autyzm, schizofrenia, neurodegeneracja), szlak NRF2.
00: 40: 27 - Zastanawiasz się nad określeniem dawki kiełków brokułów lub sulforafanu.
00: 41: 01 - Anegdoty dotyczące kiełkowania w domu.
00: 43: 14 - Przy temperaturach gotowania i aktywności sulforafanowej.
00: 43: 45 - Transformacja bakterii jelitowych sulforafanu z glukorafaniny.
00: 44: 24 - Suplementy działają lepiej w połączeniu z aktywną miozynazą z warzyw.
00: 44: 56 - Techniki gotowania i warzywa kapustne.
00: 46: 06 - Izotiocyjaniany jako goitrogeny.
Według badań naukowych Nrf2 jest podstawowym czynnikiem transkrypcyjnym, który aktywuje ochronne mechanizmy antyoksydacyjne komórek w celu detoksykacji organizmu. Jednak nadekspresja Nrf2 może powodować problemy zdrowotne. Zakres naszych informacji ogranicza się do kwestii chiropraktyki i zdrowia kręgosłupa. Aby omówić ten temat, prosimy zapytać dr Jimeneza lub skontaktować się z nami pod adresem�915-850-0900 .
Kurator: dr Alex Jimenez
Dodatkowa dyskusja na temat: Ostry ból pleców
Na ból plecówTo jedna z najczęstszych przyczyn niepełnosprawności i nieobecności w pracy na całym świecie. Ból pleców jest drugą najczęstszą przyczyną wizyt lekarskich, ustępującą jedynie infekcjom górnych dróg oddechowych. Około 80 procent populacji przynajmniej raz w życiu doświadczy bólu pleców. Kręgosłup to złożona struktura składająca się z kości, stawów, więzadeł i mięśni oraz innych tkanek miękkich. Urazy i/lub pogorszenie stanu, takie jak�przepukliny, może ostatecznie prowadzić do objawów bólu pleców. Urazy sportowe lub wypadki samochodowe są często najczęstszą przyczyną bólu pleców, jednak czasami najprostsze ruchy mogą mieć bolesne skutki. Na szczęście alternatywne opcje leczenia, takie jak opieka chiropraktyczna, mogą pomóc złagodzić ból pleców poprzez zastosowanie korekcji kręgosłupa i manualnych manipulacji, ostatecznie poprawiając ulgę w bólu.
Wiele aktualnych badań naukowych nad rakiem pozwoliło pracownikom służby zdrowia zrozumieć sposób, w jaki organizm oczyszcza się z toksyn. Analizując regulowane w górę geny w komórkach nowotworowych, naukowcy odkryli: szlak sygnałowy czynnika 2 związany z jądrowym erytroidem 2, najlepiej znany jako Nrf2. NRF2 jest ważnym czynnikiem transkrypcyjnym, który aktywuje organizm ludzki ochronne mechanizmy antyoksydacyjne w celu regulacji utleniania spowodowanego czynnikami zewnętrznymi i wewnętrznymi, aby zapobiec zwiększonemu poziomowi stresu oksydacyjnego.
Zasady Nrf2
NRF2 ma zasadnicze znaczenie dla utrzymania ogólnego stanu zdrowia i dobrego samopoczucia, ponieważ służy jako główny cel regulowania tego, jak radzimy sobie ze wszystkim, na co jesteśmy narażeni na co dzień i jak nie zachorujemy. Aktywacja NRF2 odgrywa rolę w systemie detoksykacji fazy II. Detoksykacja fazy II obejmuje lipofilowe lub rozpuszczalne w tłuszczach wolne rodniki i przekształca je w hydrofilowe lub rozpuszczalne w wodzie substancje do wydalania, jednocześnie dezaktywując wyjątkowo reaktywne metabolity i chemikalia fazy I.
Aktywacja NRF2 zmniejsza ogólne utlenianie i stany zapalne organizmu poprzez działanie hormetyczne. Aby wywołać NRF2, musi wystąpić reakcja zapalna spowodowana utlenianiem, aby komórki wytworzyły odpowiedź adaptacyjną i wytworzyły przeciwutleniacze, takie jak glutation. Aby zasadniczo przełamać zasadę Nrf2, stres oksydacyjny aktywuje NRF2, który następnie aktywuje odpowiedź antyoksydacyjną w ludzkim ciele. NRF2 działa w celu zrównoważenia sygnalizacji redoks lub równowagi poziomów utleniaczy i przeciwutleniaczy w komórce.
Doskonałą ilustrację tego, jak działa ten proces, można zademonstrować za pomocą ćwiczeń. Podczas każdego treningu mięsień dostosowuje się, aby mógł pomieścić kolejną sesję treningową. Jeśli NRF2 staje się niedostatecznie lub nadmiernie wyrażany z powodu przewlekłych infekcji lub zwiększonej ekspozycji na toksyny, co można zaobserwować u pacjentów z zespołem przewlekłej odpowiedzi zapalnej lub CIRS, problemy zdrowotne mogą się pogorszyć – po aktywacji NRF2. Przede wszystkim, jeśli DJ-1 zostanie nadmiernie utleniony, aktywacja NRF2 zakończy się zbyt szybko.
Skutki aktywacji NRF2
Aktywacja NRF2 jest silnie wyrażana w płucach, wątrobie i nerkach. Jądrowy czynnik 2 związany z erytroidem 2 lub NRF2 najczęściej działa poprzez przeciwdziałanie zwiększonemu poziomowi utleniania w organizmie człowieka, co może prowadzić do stresu oksydacyjnego. Aktywacja Nrf2 może pomóc w leczeniu różnych problemów zdrowotnych, jednak nadmierna aktywacja Nrf2 może pogorszyć różne problemy, które przedstawiono poniżej.
Okresowa aktywacja Nrf2 może pomóc:
Starzenie się (tj. Długowieczność)
Autoimmunizacja i ogólny stan zapalny (tj. zapalenie stawów, autyzm)
Rak i chemioprotekcja (tj. ekspozycja na pola elektromagnetyczne)
Depresja i lęk (tj. PTSD)
Ekspozycja na leki (alkohol, NLPZ)
Ćwiczenia i wydajność wytrzymałościowa
Choroba jelit (tj. SIBO, dysbioza, wrzodziejące zapalenie jelita grubego)
Wzrok (tj. jasne światło, wrażliwość, zaćma, dystrofia rogówki)
Hiperaktywacja Nrf2 może pogorszyć:
miażdżyca
Rak (tj. mózg, piersi, głowa, szyja, trzustka, prostata, wątroba, tarczyca)
Zespół przewlekłej odpowiedzi zapalnej (CIRS)
Przeszczep serca (podczas gdy otwarty NRF2 może być zły, NRF2 może pomóc w naprawie)
HCV
Zapalenie nerek (ciężkie przypadki)
bielactwo
Ponadto NRF2 może pomóc w działaniu określonych suplementów diety, leków i leków. Wiele naturalnych suplementów może również pomóc w wywołaniu NRF2. Dzięki obecnym badaniom naukowym naukowcy wykazali, że duża liczba związków, które kiedyś uważano za przeciwutleniacze, było naprawdę prooksydantami. To dlatego, że prawie wszystkie z nich potrzebują NRF2, nawet suplementy takie jak kurkumina i olej rybny. Na przykład wykazano, że kakao wywołuje działanie przeciwutleniające u myszy posiadających gen NRF2.
Sposoby aktywacji NRF2
W przypadku chorób neurodegeneracyjnych, takich jak choroba Alzheimera, choroba Parkinsona, udar, a nawet choroby autoimmunologiczne, prawdopodobnie najlepiej jest zwiększyć poziom Nrf2, ale w sposób hormetyczny. Mieszanie aktywatorów NRF2 może również mieć działanie addytywne lub synergistyczne, ponieważ czasami może to być zależne od dawki. Najważniejsze sposoby na zwiększenie ekspresji Nrf2 są wymienione poniżej:
HIST (ćwiczenie) + CoQ10 + słońce (te bardzo dobrze synergizują)
Kiełki brokułów + LLLT na głowie i jelitach
Maślan + Super Kawa + Poranne Słońce
Akupunktura (jest to metoda alternatywna, można również zastosować akupunkturę laserową)
Czczo
Cannabidiol (CBD)
Lwia Grzywa + Melatonina
Kwas alfa-liponowy + DIM
Piołun
Aktywacja PPAR-gamma
Poniższa obszerna lista zawierająca ponad 350 innych sposobów aktywacji Nrf2 poprzez dietę, styl życia i urządzenia, probiotyki, suplementy, zioła i oleje, hormony i neuroprzekaźniki, leki/leki i chemikalia, ścieżki/czynniki transkrypcyjne, a także inne sposoby, jest tylko krótki przewodnik na temat tego, co może wywołać Nrf2. Ze względu na zwięzłość tego artykułu pominęliśmy ponad 500 innych produktów spożywczych, suplementów diety i związków, które mogą pomóc aktywować Nrf2. Poniżej wymieniono:
Dieta:
Jagody Acai
Alkohol (czerwone wino jest lepsze, zwłaszcza jeśli jest w nim korek, ponieważ aldehyd protokatechowy z korków może również aktywować NRF2. Ogólnie rzecz biorąc, alkohol nie jest zalecany, chociaż ostre spożycie zwiększa NRF2. Przewlekłe spożycie może zmniejszać NRF2.
Glony (wodorosty morskie)
Jabłka
Czarna herbata
Orzechy brazylijskie
Kiełki Brokułów (i inne izotiocyjaniany, sulforafan, a także warzywa kapustne, takie jak bok choy, które mają D3T)
Jagody (0.6-10 g/dzień)
Marchewki (falcarinone)
Pieprz Cayenne (Kapsaicyna)
Seler (Butyloftalid)
Chaga (Betulina)
Rumiankowa herbata
Udział
Chiński Ziemniak
Aronia (Aronia)
Czekolada (Ciemna lub Kakaowa)
Cynamon
Kawa (taka jak kwas chlorogenowy, Kafestol i Kahweol)
Ćwiczenia (ostre ćwiczenia, takie jak HIST lub HIIT, wydają się być bardziej korzystne w indukowaniu NRF2, podczas gdy dłuższe ćwiczenia nie wywołują NRF2, ale zwiększają poziom glutationu)
Dieta wysokotłuszczowa (dieta)
Wysoka temperatura (sauna)
Wdychanie wodoru i woda wodorowa
Hiperbaryczna terapia tlenowa
Terapia podczerwienią (np. Joovv)
Dożylna Witamina C
Dieta ketogeniczna
Ozon
Palenie (niezalecane – ostre palenie zwiększa NRF2, chroniczne palenie zmniejsza NRF2. Jeśli zdecydujesz się na palenie, Holy Basil może pomóc chronić przed obniżoną regulacją NRF2)
Słońce (UVB i podczerwień)
Probiotyki:
Bacillus subtilis (fmbJ)
Clostridium butyricum (MIYAIRI 588)
Lactobacillus brevis
Lactobacillus casei (SC4 i 114001)
Lactobacillus collinoides
Lactobacillus gasseri (OLL2809, L13-Ia i SBT2055)
Lactobacillus helveticus (NS8)
Lactobacillus paracasei (NTU 101)
Lactobacillus plantarum (C88, CAI6, FC225, SC4)
Lactobacillus rhamnosus (GG)
Suplementy, zioła i oleje:
Acetylo-L-karnityna (ALCAR) i karnityna
Allicyny
Kwas alfa liponowy
Amentoflawon
Andrographis paniculata
agmatyny
Apigenin
Arginina
Karczoch (Cyanropicrin)
Ashwaganda
Astragalus
Bacopa
Stek Wołowy (Izogemaketon)
berberyna
Beta-karyofylen
Bidens Pilosa
Olej z nasion czarnuszki (thymoquinone)
Boswellia
Buteina
Maślan
Cannabidiol (CBD)
Karotenoidy (takie jak beta-karoten [synergia z likopenem – 2–15 mg/d likopenu], fukoksantyna, zeaksantyna, astaksantyna i luteina)
Luteolin (wysokie dawki do aktywacji, niższe dawki hamują jednak NRF2 w raku)
Magnolia
Manjistha
Maximowiczianum (Acerogenina A)
Arnika meksykańska
Ostropest
MitoQ
Mu Xiang
świerzbiec właściwy
Nikotynamid i NAD+
Panax Ginseng
Passionflower (taki jak Chrysin, ale chyryzyna może również zmniejszać NRF2 poprzez rozregulowanie sygnalizacji PI3K/Akt)
Pau d'arco (Lapacho)
Flororetin
Piceatannol
PQQ
Procyanidin
Pterostilbene
pueraria
Kwercetyna (tylko wysokie dawki, niższe dawki hamują NRF2)
Qiang Huo
Red Clover
Resweratrol (Piceid i inne fitoestrogeny zasadniczo, Rdest zwyczajny)
Rose Hips
Rosewood
Rutyna
Sapanwood
Sarsaparilla
Saururus chinensis
SC-E1 (gips, jaśmin, lukrecja, kudzu i kwiat balonowy)
Schisandra
Samoleczenie (Prunella)
Jarmułka (Bajkalin i Wogonin)
Sorrel Sheep
Si Wu Tang
Sideritis
Spikenard (Aralia)
spirulina
Ziele dziurawca
Sulforafan
Sutherlandia
Tao Hong Si Wu
Tauryna
Winorośli Boga Piorunów (Triptolide)
Tokoferole (takie jak witamina E lub linalol)
Tribulus R
Tu Si Zi
TUDCA
Witamina A (chociaż inne retinoidy hamują NRF2)
Witamina C (tylko wysoka dawka, niska dawka hamuje NRF2)
Vitex/Czyste drzewo
Biała Piwonia (Paeoniflorin z Paeonia lactiflora)
Piołun (Hispidulin i Artemisinin)
Xiao Yao Wan (wolny i łatwy wędrowiec)
Yerba Santa (Eriodictyol)
Yuan Zhi (tenuigenin)
Zi Cao (zmniejszy NRF2 w raku)
Cynk
Ziziphus Jujube
Hormony i neuroprzekaźniki:
Adiponektyna
Adropina
Estrogen (ale może zmniejszać NRF2 w tkance piersi)
Melatonina
Progesteron
Kwas chinolinowy (w odpowiedzi ochronnej zapobiegającej ekscytotoksyczności)
serotonina
Hormony tarczycy, takie jak T3 (mogą zwiększać NRF2 w zdrowych komórkach, ale zmniejszać je w przypadku raka)
Witamina D
Leki/leki i chemikalia:
Paracetamol
Acetazolamide
Amlodypina
Auranofin
Bardoksolon metylowy (BARD)
Benznidazol
BHA
CDDO-imidazolid
Ceftriakson (i antybiotyki beta-laktamowe)
Cialis
Deksametazon
Dipriwan (Propofol)
Eriodictyol
Exendin-4
ezetymib
Fluorek
Fumarate
HNE (utlenione)
Idazoxan
Arsen nieorganiczny i arsenin sodu
JQ1 (może również hamować NRF2, nieznane)
Letairis
Melphalan
metazolamid
Błękit metylenowy
Nifedypina
NLPZ
Oltipraz
PPI (takie jak Omeprazol i Lansoprazol)
Protandim – świetne wyniki in vivo, ale słabe/nieistniejące w aktywacji NRF2 u ludzi
Probucol
rapamycyna
Reserpine
Ruten
Sitaxentan
Statyny (takie jak Lipitor i Simwastatyna)
Tamoxifen
Tang Luo Ning
tBHQ
Tecfidera (fumaran dimetylu)
THC (nie tak silny jak CBD)
Teofilina
Umbelliferon
Kwas Ursodeoksycholowy (UDCA)
Werapamil
Viagra
4-acetoksyfenol
Ścieżki/czynniki transkrypcji:
?7 aktywacja nAChR?
AMPK
Bilirubina
CDK20
CKIP-1
CYP2E1
EAAT
Gankiriński
Zły duch
GJA1
ferroksydaza H-ferrytyny
Inhibitory HDAC (takie jak kwas walproinowy i TSA, ale mogą powodować niestabilność NRF2)
Białka szoku cieplnego
IL-17
IL-22
Kloto
let-7 (powala mBach1 RNA)
MAPA
Akceptanci Michaela (większość)
miR-141
miR-153
miR-155 (również niszczy mBach1 RNA)
miR-7 (w mózgu, pomaga przy nowotworach i schizofrenii)
Wycięcie1
Stres oksydacyjny (taki jak ROS, RNS, H2O2) i elektrofile
PGC-1?
PKC-delta
PPAR-gamma (efekty synergiczne)
Hamowanie receptora Sigma-1
SIRT1 (zwiększa NRF2 w mózgu i płucach, ale może go ogólnie zmniejszyć)
SIRT2
SIRT6 (w wątrobie i mózgu)
SRXN1
Inhibicja TrxR1 (również osłabienie lub wyczerpanie)
Protoporfiryna cynkowa
4-HHE
Inny:
Ankaflawina
Azbest
Awicyny
Bacillus amyloliquefaciens (stosowany w rolnictwie)
Tlenek węgla
Daphnetin
Ubytek glutationu (możliwe ubytek o 80%–90%)
gimnastyk koraiensis
HCV
Opryszczka (HSV)
Jesion indyjski
Indigowoad Root
Izosalipurpozyd
Izorhamentina
Monaszyn
Omaweloksolon (silny, znany również jako RTA-408)
PDTC
Niedobór selenu (niedobór selenu może zwiększyć NRF2)
Modrzew Syberyjski
Soforaflawanon G
Triquetrum Tadehagiego
Toona sinensis (7-DGD)
Kwiat trąbki
63171 i 63179 (silne)
Ścieżka sygnalizacyjna czynnika 2 związanego z jądrowym erytroidem 2, najlepiej znana pod skrótem Nrf2, jest czynnikiem transkrypcyjnym, który odgrywa główną rolę w regulacji ochronnych mechanizmów antyoksydacyjnych organizmu ludzkiego, szczególnie w celu kontrolowania stresu oksydacyjnego. Podczas gdy zwiększony poziom stresu oksydacyjnego może aktywować Nrf2, jego działanie jest ogromnie wzmocnione dzięki obecności określonych związków. Niektóre pokarmy i suplementy pomagają aktywować Nrf2 w ludzkim ciele, w tym sulforafan izotiocyjanianu z kiełków brokułów. Dr Alex Jimenez DC, CCST Insight
Sulforafan i jego wpływ na raka, śmiertelność, starzenie się, mózg i zachowanie, choroby serca i nie tylko
Izotiocyjaniany to jedne z najważniejszych związków roślinnych, które możesz znaleźć w swojej diecie. W tym filmie przedstawiam dla nich najbardziej wyczerpujący argument, jaki kiedykolwiek powstał. Krótki czas koncentracji? Przejdź do swojego ulubionego tematu, klikając jeden z poniższych punktów czasowych. Pełna oś czasu poniżej.
Kluczowe sekcje:
00: 01: 14 - Rak i śmiertelność
00: 19: 04 - Starzenie
00: 26: 30 - Mózg i zachowanie
00: 38: 06 - Ostatnie podsumowanie
00: 40: 27 - Dawka
Pełna oś czasu:
00: 00: 34 - Wprowadzenie sulforaphane, główny punkt widzenia wideo.
00: 01: 14 - spożycie warzyw krzyżowych i redukcja śmiertelności z wszystkich przyczyn.
00: 02: 12 - ryzyko raka prostaty.
00: 02: 23 - ryzyko raka pęcherza moczowego.
00: 02: 34 - Rak płuc w ryzyku palaczy.
00: 02: 48 - ryzyko raka piersi.
00: 03: 13 - Hipotetyczny: co, jeśli masz już raka? (interwencyjne)
00:03:35 – Prawdopodobny mechanizm napędzający dane asocjacyjne dotyczące raka i śmiertelności.
00: 04: 38 - Sulforafan i rak.
00:05:32 – Dowody na zwierzętach wykazujące silny wpływ ekstraktu z kiełków brokułów na rozwój guza pęcherza moczowego u szczurów.
00: 06: 06 - Wpływ bezpośredniej suplementacji sulforafanu u pacjentów z rakiem prostaty.
00: 11: 20 - napój z kiełków brokułów zwiększa wydalanie benzenu o 61%.
00: 13: 31 - homogenat z kiełków brokułów zwiększa enzymy antyoksydacyjne w górnych drogach oddechowych.
00: 15: 45 - śmiertelność wśród roślin krzyżowych i śmiertelność z powodu chorób serca.
00: 16: 55 - proszek z kiełków brokułów poprawia poziom lipidów we krwi i ogólne ryzyko chorób serca u chorych na cukrzycę typu 2.
00:19:04 – Początek sekcji starzenia.
00:19:21 – Dieta wzbogacona w sulforafan wydłuża życie chrząszczy od 15 do 30% (w określonych warunkach).
00: 20: 34 - Znaczenie niskiego stanu zapalnego dla długowieczności.
00: 22: 05 - warzywa kapustne i kiełki w proszku z brokułów wydają się zmniejszać szeroki zakres markerów stanu zapalnego u ludzi.
00: 23: 40 - Podsumowanie filmu wideo: rak, sekcje starzenia
00: 24: 14 - Badania na myszach sugerują, że sulforafan może poprawić zdolność adaptacyjną układu odpornościowego w starszym wieku.
00:25:18 – Sulforafan poprawił wzrost włosów w mysim modelu łysienia. Zdjęcie o 00:26:10.
00: 26: 30 - Początek sekcji mózgu i zachowania.
00: 27: 18 - Wpływ wyciągu z kiełków brokułów na autyzm.
00: 27: 48 - Wpływ glukorafaniny na schizofrenię.
00: 28: 17 - Rozpoczęcie dyskusji o depresji (prawdopodobny mechanizm i badania).
00:31:21 – Badanie na myszach przy użyciu 10 różnych modeli depresji wywołanej stresem wykazało, że sulforafan jest podobnie skuteczny jak fluoksetyna (prozac).
00: 32: 00 - Badanie pokazuje, że bezpośrednie spożycie glukorafaniny u myszy jest podobnie skuteczne w zapobieganiu depresji w modelu stresu społecznego.
00: 33: 01 - Początek sekcji zwanej neurodegeneracją.
00: 35: 01 - Sulforafan wstrzyknięty natychmiast po TBI poprawia pamięć (badanie myszy).
00: 35: 55 - Sulforafan i plastyczność neuronalna.
00:36:32 – Sulforafan poprawia uczenie się w modelu cukrzycy typu II u myszy.
00:37:19 – Dystrofia mięśniowa Sulforaphane i Duchenne'a.
00: 37: 44 - Hamowanie miostatyny w komórkach satelitarnych mięśni (in vitro).
00: 38: 06 - późne nagranie wideo: śmiertelność i rak, uszkodzenie DNA, stres oksydacyjny i zapalenie, wydalanie benzenu, choroba sercowo-naczyniowa, cukrzyca typu II, wpływ na mózg (depresja, autyzm, schizofrenia, neurodegeneracja), szlak NRF2.
00: 40: 27 - Zastanawiasz się nad określeniem dawki kiełków brokułów lub sulforafanu.
00: 41: 01 - Anegdoty dotyczące kiełkowania w domu.
00: 43: 14 - Przy temperaturach gotowania i aktywności sulforafanowej.
00: 43: 45 - Transformacja bakterii jelitowych sulforafanu z glukorafaniny.
00: 44: 24 - Suplementy działają lepiej w połączeniu z aktywną miozynazą z warzyw.
00: 44: 56 - Techniki gotowania i warzywa kapustne.
00: 46: 06 - Izotiocyjaniany jako goitrogeny.
Według wielu aktualnych badań, szlak sygnałowy czynnika 2 związanego z jądrowym erytroidem 2, najlepiej znany jako Nrf2, jest podstawowym czynnikiem transkrypcyjnym, który aktywuje ochronne mechanizmy antyoksydacyjne komórek w celu detoksykacji organizmu ludzkiego z czynników zewnętrznych i wewnętrznych oraz zapobiegania poziomy stresu oksydacyjnego. Zakres naszych informacji ogranicza się do kwestii chiropraktyki i zdrowia kręgosłupa. Aby omówić ten temat, prosimy zapytać dr Jimeneza lub skontaktować się z nami pod adresem�915-850-0900 .
Kurator: dr Alex Jimenez
Dodatkowa dyskusja na temat: Ostry ból pleców
Na ból plecówTo jedna z najczęstszych przyczyn niepełnosprawności i nieobecności w pracy na całym świecie. Ból pleców jest drugą najczęstszą przyczyną wizyt lekarskich, ustępującą jedynie infekcjom górnych dróg oddechowych. Około 80 procent populacji przynajmniej raz w życiu doświadczy bólu pleców. Kręgosłup to złożona struktura składająca się z kości, stawów, więzadeł i mięśni oraz innych tkanek miękkich. Urazy i/lub pogorszenie stanu, takie jak�przepukliny, może ostatecznie prowadzić do objawów bólu pleców. Urazy sportowe lub urazy powypadkowe są często najczęstszą przyczyną bólu pleców, jednak czasami najprostsze ruchy mogą mieć bolesne skutki. Na szczęście alternatywne opcje leczenia, takie jak chiropraktyka, mogą pomóc złagodzić ból pleców poprzez zastosowanie korekty kręgosłupa i manipulacji manualnych, ostatecznie poprawiając ulgę w bólu. �
Stres oksydacyjny jest głównym czynnikiem przyczyniającym się do rozwoju różnych problemów zdrowotnych, w tym raka, chorób serca, cukrzycy, przyspieszonego starzenia i neurodegeneracji. Pokarmy bogate w przeciwutleniacze, zioła i suplementy mogą być wykorzystywane do ochrony ludzkiego organizmu przed wysokim poziomem stresu oksydacyjnego. Ostatnie badania naukowe wykazały, że Szlak genu Nrf2 może pomóc wzmocnić działanie przeciwutleniaczy. ten zalety Nrf2 opisano poniżej.
Chroni organizm przed toksynami
NRF2 jest samoistną substancją, która może chronić komórki przed szkodliwymi, wewnętrznymi i zewnętrznymi związkami. NRF2 może pomóc wzbogacić reakcję organizmu ludzkiego na leki/leki i toksyny, poprawiając produkcję białek, które pomagają eliminować związki z komórki, znane jako białka związane z opornością wielolekową lub MRP. wdychanie dymu papierosowego, aby umożliwić detoksykację płuc.
Dodatkowo niezbędna jest ochrona płuc przed alergenami, chorobami wirusowymi, endotoksynami bakteryjnymi, hiperoksją i różnymi zanieczyszczeniami środowiska. Stały wyzwalacz Nrf2 może jednak obniżyć poziom substancji znanej jako glutation w całym ludzkim ciele. NRF2 może również chronić wątrobę przed toksycznością i może chronić wątrobę przed hepatotoksycznością arsenu. Ponadto NRF2 chroni wątrobę i mózg przed spożyciem alkoholu. Na przykład Nrf2 może chronić przed toksycznością paracetamolu.
Zwalcza stany zapalne i stres oksydacyjny
Aktywacja NRF2 może pomóc w walce z zapaleniem poprzez zmniejszenie zapalnych cytokin, takich jak te obecne w łuszczycy. NRF2 może również zmniejszać stan zapalny związany z różnymi problemami zdrowotnymi, takimi jak zapalenie stawów i zwłóknienie wątroby, nerek i płuc. NRF2 może również pomóc w kontrolowaniu alergii poprzez obniżenie poziomu cytokin Th1/Th17 i podniesienie poziomu cytokin TH2. Może to być korzystne w przypadku dolegliwości takich jak astma.
NRF2 dodatkowo chroni przed uszkodzeniem komórek przez światło niebieskie i promieniowanie UVA/UVB występujące w świetle słonecznym. Niedobory Nrf2 mogą znacznie ułatwić oparzenie słoneczne. Jednym z powodów tego jest fakt, że NRF2 jest w stanie regulować kolagen w odpowiedzi na promieniowanie UV. Produkty końcowe zaawansowanej glikacji lub AGE przyczyniają się do rozwoju wielu problemów zdrowotnych, w tym cukrzycy i chorób neurodegeneracyjnych. NRF2 może zmniejszyć stres oksydacyjny AGE w organizmie. NRF2 może również chronić ludzkie ciało przed wyższym poziomem stresu związanego z ciepłem.
Poprawia mitochondria i wydajność ćwiczeń
NRF2 to wzmacniacz mitochondrialny. Aktywacja NRF2 przyczynia się do wzrostu energii ATP dla mitochondriów, oprócz zwiększonego wykorzystania tlenu lub cytrynianu i tłuszczu. Bez NRF2 mitochondria mogłyby po prostu funkcjonować z cukrem lub glukozą, a nie z tłuszczem. NRF2 jest również niezbędny do rozwoju mitochondriów w procesie znanym jako biogeneza. Aktywacja NRF2 jest niezbędna, aby „wykorzystać” korzyści płynące z ćwiczeń.
Ze względu na aktywność Nrf2, ćwiczenia poprawiają funkcję mitochondriów, gdzie wynik ten może być wzmocniony przez CoQ10, Cordyceps i ograniczenie kalorii. Umiarkowany wysiłek fizyczny lub intensywny wysiłek fizyczny indukują biogenezę mitochondriów i podwyższoną syntezę dysmutazy ponadtlenkowej (SOD) i hemeoksygenazy-1 (HO-1), poprzez aktywację NRF2. Kwas alfa-liponowy lub ALA i Dan Shen mogą zwiększyć biogenezę mitochondriów za pośrednictwem NRF2. Co więcej, NRF2 może również poprawić tolerancję wysiłku, gdy delecja NRF2 powoduje, że ćwiczenia są szkodliwe.
Chroni przed niedotlenieniem
NRF2 pomaga również chronić organizm ludzki przed utratą/zubożeniem tlenu w komórkach, problemem zdrowotnym zwanym niedotlenieniem. Osoby z CIRS mają obniżony poziom tlenu, ponieważ ich NRF2 jest zablokowany, co skutkuje obniżonymi poziomami zarówno VEGF, HIF1, jak i HO-1. Zwykle u zdrowych osób z niedotlenieniem miR-101, który jest wymagany do tworzenia komórek macierzystych, ulega nadekspresji i zwiększa ilość NRF2/HO-1 i VEGF/eNOS, zapobiegając w ten sposób uszkodzeniom mózgu, ale wydaje się, że nie występuje w CIRS.
Niedotlenienie, charakteryzujące się niskim HIF1 w CIRS, może również skutkować nieszczelną barierą krew-mózg z powodu braku równowagi NRF2. Salidroside, znajdujący się w Rhodiola, działa na aktywację NRF2 i pomaga w niedotlenieniu poprzez zwiększenie poziomu VEGF i HIF1 w ludzkim ciele. NRF2 może również ostatecznie chronić przed gromadzeniem się mleczanu w sercu. Aktywacja NRF2 może również zatrzymać wywołaną niedotlenieniem chorobę wysokościową lub AMS.
Spowalnia starzenie
Kilka związków, które mogą być śmiertelne w ogromnych ilościach, może wydłużyć żywotność w niewielkich ilościach z powodu ksenohormezy przez NRF2, PPAR-gamma i FOXO. Bardzo mała ilość toksyn zwiększa zdolność komórki do lepszego przygotowania do następnego kontaktu z toksyną, jednak nie jest to zachęta do spożywania trujących chemikaliów.
Dobrą ilustracją tego procesu jest ograniczenie kalorii. NRF2 może wydłużyć żywotność komórek, podnosząc ich poziom mitochondriów i przeciwutleniaczy, a także obniżając zdolność komórek do śmierci. NRF2 zmniejsza się wraz z wiekiem, ponieważ NRF2 zapobiega obumieraniu komórek macierzystych i wspomaga ich regenerację. NRF2 odgrywa rolę w przyspieszaniu gojenia się ran.
Wzmacnia układ naczyniowy
Aktywacja NRF2 przeprowadzona prawidłowo z produkcją sulforafanu może chronić przed chorobami serca, takimi jak wysokie ciśnienie krwi lub nadciśnienie, stwardnienie tętnic lub miażdżyca. NRF2 może wzmacniać relaksującą aktywność acetylocholiny lub ACh w układzie naczyniowym, jednocześnie zmniejszając stres wywołany przez cholesterol. Aktywacja Nrf2 może wzmacniać serce, jednak nadmierna aktywacja Nrf2 może zwiększać prawdopodobieństwo wystąpienia choroby sercowo-naczyniowej.
Statyny mogą zapobiegać chorobom układu krążenia lub prowadzić do nich. NRF2 odgrywa również ważną rolę w równoważeniu żelaza i wapnia, które mogą chronić ludzkie ciało przed podwyższonym poziomem żelaza. Przykładowo, Sirtuina 2 lub SIRT2 może regulować homeostazę żelaza w komórkach poprzez aktywację NRF2, która, jak się uważa, jest wymagana dla zdrowych poziomów żelaza. NRF2 może również pomóc w przypadku niedokrwistości sierpowatej lub SCD. Dysfunkcja NRF2 może być przyczyną endotoksemii, podobnie jak w przypadku dysbiozy czy nadciśnienia indukowanego lektynami. Nrf2 może również chronić organizm przed uszkodzeniem układu naczyniowego wywołanym przez amfetaminę.
Zwalcza zapalenie nerwów
NRF2 może chronić i wspomagać zapalenie mózgu, powszechnie określane jako neurozapalenie. Ponadto NRF2 może pomóc w asortymencie zaburzeń ośrodkowego układu nerwowego (OUN), w tym:
Choroba Alzheimera (AD) – zmniejsza stres beta-amyloidowy w mitochondriach
Stwardnienie zanikowe boczne (ALS)
Choroba Huntingtona (HD)
Stwardnienie rozsiane (MS)
Regeneracja nerwów
choroba Parkinsona (PD) – chroni dopaminę
Uraz rdzenia kręgowego (SCI)
Udar (niedokrwienny i krwotoczny) – wspomaga niedotlenienie
Urazie mózgu
NRF2 ujawnił zmniejszenie neurozapalenia u nastolatków z zaburzeniami ze spektrum autyzmu lub ASD. Idebenone prawidłowo paruje się z aktywatorami NRF2 w przeciwieństwie do zapalenia układu nerwowego. NRF2 może również poprawić barierę krew-mózg lub BBB. Przykładowo, aktywacja NRF2 kwasem karnozowym uzyskanym z rozmarynu i szałwii może przekroczyć BBB i wywołać neurogenezę. Wykazano również, że NRF2 podnosi czynnik neurotroficzny pochodzenia mózgowego (BDNF).
NRF2 moduluje również zdolność niektórych suplementów diety do wywoływania czynnika wzrostu nerwów lub NGF, ponieważ może również pomóc w problemach wywołanych mgłą mózgową i glutaminianem poprzez modulowanie N-metylo-D-asparaginianu lub receptorów NMDA. Może również obniżać stres oksydacyjny wywołany kwasem chinolinowym, określanym jako QUIN. Aktywacja NRF2 może chronić przed napadami, a duże dawki mogą zmniejszyć granicę napadu. Przy regularnych dawkach stymulacji, NRF2 może zwiększać zdolności poznawcze po napadach padaczkowych poprzez obniżanie pozakomórkowego glutaminianu w mózgu i poprzez zdolność do pobierania cysteiny z glutaminianu i glutationu.
Łagodzi depresję
W depresji normalne jest zauważenie stanu zapalnego w mózgu, zwłaszcza kory przedczołowej i hipokampa, a także obniżonego BDNF. W niektórych wersjach depresji NRF2 może złagodzić objawy depresji poprzez zmniejszenie stanu zapalnego w mózgu i zwiększenie poziomu BDNF. Zdolność agmatyny do zmniejszania depresji poprzez podnoszenie noradrenaliny, dopaminy, serotoniny i BDNF w hipokampie zależy od aktywacji NRF2.
Zawiera właściwości przeciwnowotworowe
NRF2 jest zarówno supresorem nowotworu, jak i promotorem nowotworu, jeśli nie jest odpowiednio leczony. NRF2 może chronić przed rakiem wywołanym przez wolne rodniki i stres oksydacyjny, jednak nadekspresję NRF2 można również znaleźć w komórkach rakowych. Intensywna aktywacja NRF2 może pomóc w przypadku różnych nowotworów. Na przykład suplement Protandim może zmniejszyć raka skóry poprzez aktywację NRF2.
Uśmierza ból
Gulf War Illness lub GWI, godna uwagi choroba dotykająca weteranów wojny w Zatoce, to zbiór niewyjaśnionych, przewlekłych objawów, które mogą obejmować zmęczenie, bóle głowy, bóle stawów, niestrawność, bezsenność, zawroty głowy, dolegliwości oddechowe i problemy z pamięcią. NRF2 może złagodzić objawy GWI poprzez zmniejszenie hipokampa i ogólnego stanu zapalnego, a także zmniejszenie bólu. NRF2 może dodatkowo wspomagać ból spowodowany uszkodzeniem nerwów ciała i poprawiać uszkodzenie nerwów spowodowane neuropatią cukrzycową.
Poprawia cukrzycę
Wysoki poziom glukozy, najlepiej określany jako hiperglikemia, powoduje uszkodzenie oksydacyjne komórek z powodu zaburzenia funkcji mitochondriów. Aktywacja NRF2 może chronić organizm ludzki przed uszkodzeniem komórki przez hiperglikemię, zapobiegając w ten sposób śmierci komórki. Aktywacja NRF2 może dodatkowo chronić, przywracać i wzmacniać funkcję komórek beta trzustki, jednocześnie zmniejszając insulinooporność.
Chroni wzrok i słuch
NRF2 może chronić oko przed retinopatią cukrzycową. Może również zapobiegać powstawaniu zaćmy i chronić fotoreceptory w przeciwieństwie do śmierci wywołanej światłem. NRF2 dodatkowo chroni ucho lub ślimak przed stresem i utratą słuchu.
Może pomóc w otyłości
NRF2 może pomóc w otyłości przede wszystkim ze względu na jego zdolność do regulowania zmiennych, które wpływają na gromadzenie tłuszczu w ludzkim ciele. Aktywacja NRF2 za pomocą sulforafanu może zwiększyć hamowanie syntezy kwasów tłuszczowych (FAS) i białek rozprzęgających (UCP), co skutkuje mniejszą akumulacją tłuszczu i większą ilością brązowego tłuszczu, określanego jako tłuszcz, który zawiera więcej mitochondriów.
Chroni jelita
NRF2 pomaga chronić jelita, chroniąc homeostazę mikrobiomu jelitowego. Na przykład probiotyki Lactobacillus będą wyzwalać NRF2 do ochrony jelit przed stresem oksydacyjnym. NRF2 może również pomóc w zapobieganiu wrzodziejącemu zapaleniu jelita grubego lub UC.
Chroni narządy płciowe
NRF2 może chronić jądra i chronić liczbę plemników przed uszkodzeniem u osób z cukrzycą. Może również pomóc w zaburzeniach erekcji lub ED. Niektóre suplementy zwiększające libido, takie jak Mucuna, Tribulus i Ashwaganda, mogą poprawiać funkcje seksualne poprzez aktywację NRF2. Inne czynniki, które zwiększają NRF2, takie jak światło słoneczne lub kiełki brokułów, mogą również pomóc w poprawie libido.
Reguluje kości i mięśnie
Stres oksydacyjny może powodować zmniejszenie gęstości i siły kości, co jest normalne w osteoporozie. Aktywacja NRF2 może mieć zdolność do poprawy przeciwutleniaczy w kościach i ochrony przed starzeniem się kości. NRF2 może również zapobiegać utracie mięśni i wzmacniać dystrofię mięśniową Duchenne'a lub DMD.
Zawiera właściwości antywirusowe
Wreszcie, aktywacja NRF2 może ostatecznie pomóc w obronie organizmu ludzkiego przed kilkoma wirusami. U pacjentów z wirusem dengi objawy nie były tak nasilone u osób, które miały wyższy poziom NRF2 w porównaniu z osobami, które miały niższy poziom NRF2. NRF2 może również pomóc osobom z wirusem ludzkiego niedoboru odporności-1 lub HIV. NRF2 może chronić przed stresem oksydacyjnym wywołanym przez wirusy adenowirusowe (AAV) i H. Pylori. Wreszcie, korzeń Lindera może tłumić wirusa zapalenia wątroby typu C poprzez aktywację NRF2.
Nrf2 lub czynnik 2 związany z NF-E2 jest czynnikiem transkrypcyjnym występującym u ludzi, który reguluje ekspresję określonego zestawu genów antyoksydacyjnych i detoksykujących. Ta ścieżka sygnalizacyjna jest aktywowana z powodu stresu oksydacyjnego, ponieważ wzmacnia liczne enzymy antyoksydacyjne i detoksykacyjne wątroby fazy II w celu przywrócenia homeostazy w ludzkim ciele. Ludzie są przystosowani do funkcjonowania w stanie homeostazy lub równowagi. Kiedy organizm jest skonfrontowany ze stresem oksydacyjnym, Nrf2 aktywuje się, aby regulować utlenianie i kontrolować stres, który powoduje. Nrf2 jest niezbędny do zapobiegania problemom zdrowotnym związanym ze stresem oksydacyjnym. Dr Alex Jimenez DC, CCST Insight
Sulforafan i jego wpływ na raka, śmiertelność, starzenie się, mózg i zachowanie, choroby serca i nie tylko
Izotiocyjaniany to jedne z najważniejszych związków roślinnych, które możesz znaleźć w swojej diecie. W tym filmie przedstawiam dla nich najbardziej wyczerpujący argument, jaki kiedykolwiek powstał. Krótki czas koncentracji? Przejdź do swojego ulubionego tematu, klikając jeden z poniższych punktów czasowych. Pełna oś czasu poniżej.
Kluczowe sekcje:
00: 01: 14 - Rak i śmiertelność
00: 19: 04 - Starzenie
00: 26: 30 - Mózg i zachowanie
00: 38: 06 - Ostatnie podsumowanie
00: 40: 27 - Dawka
Pełna oś czasu:
00: 00: 34 - Wprowadzenie sulforaphane, główny punkt widzenia wideo.
00: 01: 14 - spożycie warzyw krzyżowych i redukcja śmiertelności z wszystkich przyczyn.
00: 02: 12 - ryzyko raka prostaty.
00: 02: 23 - ryzyko raka pęcherza moczowego.
00: 02: 34 - Rak płuc w ryzyku palaczy.
00: 02: 48 - ryzyko raka piersi.
00: 03: 13 - Hipotetyczny: co, jeśli masz już raka? (interwencyjne)
00:03:35 – Prawdopodobny mechanizm napędzający dane asocjacyjne dotyczące raka i śmiertelności.
00: 04: 38 - Sulforafan i rak.
00:05:32 – Dowody na zwierzętach wykazujące silny wpływ ekstraktu z kiełków brokułów na rozwój guza pęcherza moczowego u szczurów.
00: 06: 06 - Wpływ bezpośredniej suplementacji sulforafanu u pacjentów z rakiem prostaty.
00: 11: 20 - napój z kiełków brokułów zwiększa wydalanie benzenu o 61%.
00: 13: 31 - homogenat z kiełków brokułów zwiększa enzymy antyoksydacyjne w górnych drogach oddechowych.
00: 15: 45 - śmiertelność wśród roślin krzyżowych i śmiertelność z powodu chorób serca.
00: 16: 55 - proszek z kiełków brokułów poprawia poziom lipidów we krwi i ogólne ryzyko chorób serca u chorych na cukrzycę typu 2.
00:19:04 – Początek sekcji starzenia.
00:19:21 – Dieta wzbogacona w sulforafan wydłuża życie chrząszczy od 15 do 30% (w określonych warunkach).
00: 20: 34 - Znaczenie niskiego stanu zapalnego dla długowieczności.
00: 22: 05 - warzywa kapustne i kiełki w proszku z brokułów wydają się zmniejszać szeroki zakres markerów stanu zapalnego u ludzi.
00: 23: 40 - Podsumowanie filmu wideo: rak, sekcje starzenia
00: 24: 14 - Badania na myszach sugerują, że sulforafan może poprawić zdolność adaptacyjną układu odpornościowego w starszym wieku.
00:25:18 – Sulforafan poprawił wzrost włosów w mysim modelu łysienia. Zdjęcie o 00:26:10.
00: 26: 30 - Początek sekcji mózgu i zachowania.
00: 27: 18 - Wpływ wyciągu z kiełków brokułów na autyzm.
00: 27: 48 - Wpływ glukorafaniny na schizofrenię.
00: 28: 17 - Rozpoczęcie dyskusji o depresji (prawdopodobny mechanizm i badania).
00:31:21 – Badanie na myszach przy użyciu 10 różnych modeli depresji wywołanej stresem wykazało, że sulforafan jest podobnie skuteczny jak fluoksetyna (prozac).
00: 32: 00 - Badanie pokazuje, że bezpośrednie spożycie glukorafaniny u myszy jest podobnie skuteczne w zapobieganiu depresji w modelu stresu społecznego.
00: 33: 01 - Początek sekcji zwanej neurodegeneracją.
00: 35: 01 - Sulforafan wstrzyknięty natychmiast po TBI poprawia pamięć (badanie myszy).
00: 35: 55 - Sulforafan i plastyczność neuronalna.
00:36:32 – Sulforafan poprawia uczenie się w modelu cukrzycy typu II u myszy.
00:37:19 – Dystrofia mięśniowa Sulforaphane i Duchenne'a.
00: 37: 44 - Hamowanie miostatyny w komórkach satelitarnych mięśni (in vitro).
00: 38: 06 - późne nagranie wideo: śmiertelność i rak, uszkodzenie DNA, stres oksydacyjny i zapalenie, wydalanie benzenu, choroba sercowo-naczyniowa, cukrzyca typu II, wpływ na mózg (depresja, autyzm, schizofrenia, neurodegeneracja), szlak NRF2.
00: 40: 27 - Zastanawiasz się nad określeniem dawki kiełków brokułów lub sulforafanu.
00: 41: 01 - Anegdoty dotyczące kiełkowania w domu.
00: 43: 14 - Przy temperaturach gotowania i aktywności sulforafanowej.
00: 43: 45 - Transformacja bakterii jelitowych sulforafanu z glukorafaniny.
00: 44: 24 - Suplementy działają lepiej w połączeniu z aktywną miozynazą z warzyw.
00: 44: 56 - Techniki gotowania i warzywa kapustne.
00: 46: 06 - Izotiocyjaniany jako goitrogeny.
Kiedy organizm ludzki zostaje skonfrontowany ze szkodliwymi czynnikami wewnętrznymi i zewnętrznymi, takimi jak toksyny, komórki muszą szybko uruchomić swoje zdolności antyoksydacyjne, aby przeciwdziałać stresowi oksydacyjnemu. Ponieważ stwierdzono, że podwyższony poziom stresu oksydacyjnego powoduje różne problemy zdrowotne, ważne jest, aby użyć aktywacji Nrf2, aby skorzystać z jej zalet. Zakres naszych informacji ogranicza się do kwestii chiropraktyki i zdrowia kręgosłupa. Aby omówić ten temat, prosimy zapytać dr Jimeneza lub skontaktować się z nami pod adresem�915-850-0900 .
Kurator: dr Alex Jimenez
Dodatkowa dyskusja na temat: Ostry ból pleców
Na ból plecówTo jedna z najczęstszych przyczyn niepełnosprawności i opuszczonych dni w pracy na całym świecie. Ból pleców to druga najczęstsza przyczyna wizyt lekarskich, przewyższająca liczebnie jedynie infekcje górnych dróg oddechowych. Około 80 procent populacji przynajmniej raz doświadczy bólu pleców w ciągu swojego życia. Kręgosłup to złożona struktura składająca się z kości, stawów, więzadeł i mięśni oraz innych tkanek miękkich. Z tego powodu urazy i / lub zaostrzone warunki, takie jak przepukliny, może ostatecznie prowadzić do objawów bólu pleców. Urazy sportowe lub urazy powypadkowe są często najczęstszą przyczyną bólu pleców, jednak czasami najprostsze ruchy mogą mieć bolesne skutki. Na szczęście alternatywne opcje leczenia, takie jak chiropraktyka, mogą pomóc złagodzić ból pleców poprzez zastosowanie korekty kręgosłupa i manipulacji manualnych, ostatecznie poprawiając ulgę w bólu. �
Sulforafan jest substancją fitochemiczną, substancją z grupy izotiocyjanianowej związków siarkoorganicznych, występującą w warzywach kapustowatych, takich jak brokuły, kapusta, kalafior i brukselka. Można go również znaleźć w bok choy, jarmuż, collardach, musztardach i rzeżuchy. Badania naukowe wykazały, że sulforafan może pomóc w profilaktyce różnych typów raka aktywacja produkcji Nrf2lub czynnik jądrowy związany z czynnikiem erytroidalnym 2, czynnik transkrypcyjny, który reguluje ochronne mechanizmy przeciwutleniające, które kontrolują odpowiedź komórki na utleniacze. Celem poniższego artykułu jest opisanie funkcji sulforafanu.
Abstrakcyjny
System przeciwutleniający KEAP1-Nrf2-ARE jest głównym środkiem, dzięki któremu komórki reagują na stres oksydacyjny i ksenobiotyczny. Sulforafan (SFN), elektrofilowy izotiocyjanian pochodzący z roślin krzyżowych, aktywuje szlak KEAP1-Nrf2-ARE i stał się cząsteczką będącą przedmiotem zainteresowania w leczeniu chorób, w których przewlekły stres oksydacyjny odgrywa główną rolę etiologiczną. Pokazujemy tutaj, że mitochondria hodowanych komórek nabłonka barwnikowego siatkówki (RPE-1) traktowanych SFN ulegają hiperfuzji, która jest niezależna od zarówno Nrf2, jak i jego inhibitora cytoplazmatycznego KEAP1. Doniesiono, że fuzja mitochondrialna jest cytoochronna poprzez hamowanie tworzenia się porów w mitochondriach podczas apoptozy i zgodnie z tym, pokazujemy niezależną od NrXXUM, cytoprotekcję komórek traktowanych SFN eksponowanych na induktor apoptozy, staurosporynę. Mechanicznie, SFN łagodzi rekrutację i / lub zatrzymywanie rozpuszczalnego czynnika rozszczepienia Drp2 do mitochondriów i peroksysomów, ale nie wpływa na ogólną obfitość Drp1. Dane te pokazują, że korzystne właściwości SFN wykraczają poza aktywację systemu KEAP1-Nrf1-ARE i uzasadniają dalsze przesłuchania, biorąc pod uwagę obecne zastosowanie tego środka w wielu badaniach klinicznych.
Słowa kluczowe:Sulforafan, Nrf2, Drp1, mitochondria, rozszczepienie, fuzja, apoptoza
Wprowadzenie
Sulforafan jest niezależnym inhibitorem rozszczepienia mitochondrialnego Nrf2
Sulforafan (SFN) to związek izotiocyjanianowy powstały w diecie najczęściej z warzyw kapustnych [56]. Jest wytwarzany w roślinach jako ksenobiotyczna odpowiedź na drapieżnictwo poprzez pęcherzykowe uwalnianie enzymu hydrolitycznego miozynazy z uszkodzonych komórek; ten enzym przekształca glukozynolany w izotiocyjaniany [42]. W ciągu ostatnich dwóch dekad SFN została szeroko scharakteryzowana ze względu na zgłoszone właściwości przeciwnowotworowe, przeciwutleniające i przeciwdrobnoustrojowe [57]. Znaczna część tej skuteczności została przypisana zdolności SFN do modulowania szlaku sygnałowego elementu odpowiedzi przeciwutleniającej KEAP1-Nrf2 (ARE), chociaż zidentyfikowano dodatkowe aktywności związku, w tym hamowanie aktywności deacetylazy histonowej i progresję cyklu komórkowego [ 29]. Nrf2 jest głównym czynnikiem transkrypcyjnym antyoksydacyjnym, a w warunkach homeostazy jego stabilność jest tłumiona przez działanie kompleksu ligazy ubikwitynowej Cullin3KEAP1 [20]. W szczególności, Nrf2 jest rekrutowany do ligazy Cullin3KEAP1 przez wiązanie z dimerycznym adapterem substratu KEAP1 i jest następnie modyfikowany łańcuchami poliUb, których celem jest czynnik transkrypcyjny dla degradacji za pośrednictwem proteasomu. Ten konstytutywny obrót ogranicza czas trwania Nrf2 w komórkach bez stresu do ~ 15 min [30], [33], [46], [55]. W odpowiedzi na wiele rodzajów stresu, w szczególności stres oksydacyjny, KEAP1, białko bogate w cysteinę, działa jako czujnik redoks, a oksydacyjna modyfikacja krytycznych cystein, w szczególności C151, KEAP1 dysocjuje Nrf2-KEAP1 z CUL3, zapobiegając w ten sposób degradacji Nrf2 [ 8], [20], [55]. Warto zauważyć, że SFN i prawdopodobnie inne aktywatory Nrf2 naśladują stres oksydacyjny poprzez modyfikację C151 z KEAP1 np. [21]. Stabilizacja Nrf2 pozwala na jego translokację do jądra, gdzie indukuje ekspresję baterii genów antyoksydantów i detoksykacji II fazy. Nrf2 wiąże się z elementami promotora odpowiedzi antyoksydacyjnej (ARE) pokrewnych genów docelowych poprzez heterodimeryzację z małymi białkami Maf [19]. System ten wykazuje dynamiczną i wrażliwą reakcję na pośrednie przeciwutleniacze, takie jak SFN, wolne rodniki generowane przez mitochondria [16] lub inne fizjologiczne źródła stresu oksydacyjnego [41].
Mitochondria są dynamicznymi, subkomórkowymi organellami, które regulują wiele funkcji komórkowych, od produkcji ATP i wewnątrzkomórkowego buforowania wapnia do regulacji redoks i apoptozy [13], [49]. Mitochondria stanowią również główne źródło reaktywnych form tlenu (ROS) w komórce. Właściwa regulacja funkcji mitochondrialnych jest zatem konieczna do optymalizacji produkcji ATP w celu zaspokojenia potrzeb komórkowych, przy jednoczesnym minimalizowaniu potencjalnie szkodliwych skutków nadmiernej produkcji wolnych rodników. Krytycznym wymogiem dla dobrej modulacji funkcji mitochondriów jest zdolność mitochondriów do funkcjonowania zarówno niezależnie, jak maszyny biochemiczne i jako część rozległej, reagującej sieci.
Morfologia i funkcja sieci mitochondrialnej są określone przez regulowaną równowagę między rozszczepieniem a stopniem zespolenia. Rozszczepienie mitochondrialne jest wymagane do dziedziczenia mitochondriów przez komórki potomne podczas podziału komórki [28], jak również do selektywnej, autofagicznej degradacji depolaryzowanych lub uszkodzonych mitochondriów, określanych jako mitofagia [1]. Odwrotnie, fuzja jest wymagana do komplementacji genomów mitochondrialnych i współdzielenia łańcuchów transportu elektronów pomiędzy sąsiednimi mitochondriami [54]. Na poziomie molekularnym, rozszczepienie mitochondriów i fuzja są regulowane przez duże, GINazy typu dynaminowego. Trzy enzymy głównie regulują fuzję: Mitofusins 1 i 2 (Mfn1 / 2) są dwuprzebiegowymi białkami błony zewnętrznej, które pośredniczą w fuzji błony zewnętrznej poprzez oddziaływania heterotypowe między sąsiadującymi mitochondriami [15], [25], [37], podczas gdy OPA1 jest wewnętrznym białko błonowe, które jednocześnie zapewnia łączność macierzową poprzez regulację łączenia błon wewnętrznych [5]. Aktywność GTPazy wszystkich trzech białek jest wymagana do stabilnej fuzji [5], [18], a OPA1 jest dalej regulowany przez kompleksową proteolizę w wewnętrznej błonie mitochondrialnej przez proteazy OMA1 [14], PARL [6] i YME1L [45 ]. Co ważne, do prawidłowej fuzji niezbędny jest nienaruszony potencjał błonowy mitochondriów w celu powstrzymania integracji uszkodzonych i zdrowych mitochondriów [26].
Rozszczepienie mitochondrialne jest głównie katalizowane przez białko cytozolowe zwane białkiem 1 (Drp1 / DNM1L). Drp1 jest rekrutowany z cytozolu do potencjalnych miejsc rozszczepienia na zewnętrznej błonie mitochondrialnej [43]. Głównymi receptorami Drp1 na zewnętrznej błony są mitochondrialny czynnik rozszczepienia (Mff) [32] i, w mniejszym stopniu, Fission 1 (Fis1) [51]. Dodatkowo odkryto receptor wabika, MIEF1 / MiD51, który działa w celu dalszego ograniczenia aktywności białka Drp1 w potencjalnych miejscach rozszczepienia [58]. Po przyłączeniu do zewnętrznej błony mitochondrialnej Drp1 oligomeryzuje do struktur spiralnych wokół ciała mitochondrium, a następnie wykorzystuje energię pochodzącą z hydrolizy GTP do pośredniczenia w fizycznym rozkładzie mitochondrialnych zewnętrznych i wewnętrznych błon [17]. Kanaliki pochodzące z retikulum endoplazmatycznego działają jako początkowe drenowanie mitochondriów przed oligomeryzacją Drp1, co podkreśla odkrycie, że nieskrępowane mitochondria są szersze niż obwód permisywny ukończonej spirali Drp1 [12]. Dynamika aktyn jest również ważna dla interakcji mitochondriów ER, które poprzedzają podział mitochondrialny [24]. Oprócz swojej roli w rozszczepieniu mitochondriów, Drp1 katalizuje rozszczepienie peroksysomów [40].
Drp1 jest bardzo podobny do dobrze scharakteryzowanego białka dynaminy, ponieważ oba białka zawierają N-końcową domenę GTPase, domenę środkową, która jest krytyczna dla samo-oligomeryzacji i C-końcową domenę efektorową GTPazy [31]. Drp1 osiąga selektywność dla błon mitochondrialnych poprzez połączenie interakcji z białkami receptorowymi Mff i Fis1, a także dzięki swojemu powinowactwu do kardiolipiny fosfolipidowej specyficznej dla mitochondriów poprzez unikalną domenę wstawki B Drp1 [2]. Drp1 zazwyczaj istnieje jako homotetramer w cytoplazmie, a do składania wyższych rzędów w mitochondrialnych miejscach rozszczepienia pośredniczy domena Drp1 [3].
Biorąc pod uwagę ukryty związek między funkcjonowaniem mitochondriów a szlakiem KEAP1-Nrf2-ARE, badaliśmy wpływ aktywacji Nrf2 na strukturę i funkcję mitochondrium. Pokazujemy tutaj, że SFN indukuje hiperfuzję mitochondrialną, która nieoczekiwanie jest niezależna od zarówno Nrf2, jak i KEAP1. Ten efekt SFN polega na hamowaniu funkcji Drp1. Dalej wykazujemy, że SFN nadaje oporność na apoptozę niezależną od Nrf2 i naśladuje tę obserwowaną w komórkach zubożonych Drp1. Dane te łącznie wskazują, że oprócz stabilizowania i aktywacji Nrf2, SFN moduluje dynamikę mitochondrialną i zachowuje sprawność i przetrwanie komórek.
W trakcie badania wpływu aktywacji Nrf2 na dynamikę sieci mitochondrialnej odkryliśmy, że traktowanie unieśmiertelnionych ludzkich komórek nabłonka barwnikowego siatkówki (RPE-1) sulforafanem (SFN), silnym aktywatorem sygnalizacji Nrf2, indukuje silną fuzję sieć mitochondrialna w porównaniu z komórkami kontrolnymi traktowanymi nośnikiem (ryc. 1A i B). Morfologia mitochondriów w tych komórkach bardzo przypominała mitochondria w komórkach zubożonych przez siRNA endogennego Drp1, głównego czynnika rozszczepienia mitochondriów (ryc. 1A). Wynik ten zrodził intrygujący pomysł, że rozszczepienie mitochondriów i stan fuzji odpowiadają bezpośrednio na poziom Nrf2 w komórce. Jednak stymulacja komórek innymi stabilizatorami i aktywatorami Nrf2, takimi jak inhibitor proteasomu MG132, prooksydant tBHQ lub knockdown inhibitora Nrf2 KEAP1 nie indukowały fuzji mitochondrialnej (ryc. 1A i B). Stabilizacja Nrf2 przez te manipulacje została potwierdzona metodą western blot dla endogennego Nrf2 (Fig. 1C). Ponadto ekspresja Nrf2 była zbędna w przypadku fuzji mitochondrialnej indukowanej przez SFN, ponieważ knockdown endogennego Nrf2 przez siRNA nie przeciwdziała temu fenotypowi (ryc. 1D F). Ponieważ SFN stymuluje szlak KEAP1-Nrf2-ARE poprzez kowalencyjną modyfikację reszt cysteiny w KEAP1 [21], znokautowaliśmy KEAP1, aby sprawdzić, czy indukowana SFN hiperfuzja mitochondrialna jest stymulowana przez szlak zależny od KEAP1, ale niezależny od Nrf2. Jednak zubożenie KEAP1 również nie zdołało znieść fuzji mitochondrialnej indukowanej przez SFN (ryc. 1G I). W rzeczywistości SFN odwrócił morfologię pryzmatyczną indukowaną przez zubożenie KEAP1 (ryc. 1G, panel b w porównaniu z panelem d). Wyniki te wskazują, że leczenie SFN powoduje fuzję mitochondriów niezależnie od kanonicznego szlaku KEAP1-Nrf2-ARE i doprowadziło nas do zbadania, czy SFN bezpośrednio wpływa na elementy rozszczepienia mitochondriów lub maszynerii fuzji.
Figura 1 SFN indukuje niezależną fuzję mitochondrialną Nrf2 / KEAP1. (A) Komórki RPE-1 transfekowano wskazanymi siRNA i 3 dni później traktowano DMSO lub aktywatorami Nrf2 SFN (50 µM), MG132 (10 µM) lub tBHQ (100 µM) przez 4 godziny. Mitochondria (czerwone) są znakowane przeciwciałem anty-Tom20, a jądra (niebieskie) są barwione kontrastowo za pomocą DAPI. (B) Wykres przedstawiający kwantyfikację oceny morfologii mitochondriów na podstawie (A). > 50 komórek na stan oceniano w sposób zaślepiony. (C) Reprezentatywne western bloty z (A). (D) Komórki RPE-1 transfekowano 10 nM siRNA i 3 dni później traktowano SFN przez 4 godziny przed utrwaleniem i wybarwieniem jak w (A). (E) Wykres przedstawiający kwantyfikację oceny fenotypu mitochondrialnego na podstawie (D). > 100 komórek na stan oceniano w sposób zaślepiony. (F) Reprezentatywne western bloty z (D). (G) Komórki transfekowano i traktowano jak w (D) siCON lub siKEAP1. (H) Komórki z (G) oceniano jak w (B) i (E) na podstawie morfologii mitochondriów. (I) Reprezentatywne western bloty z (G). Dane w (B), (E) i (H) zebrano z 3 niezależnych eksperymentów, z których każdy i istotność statystyczną określono za pomocą dwustronnego testu t-Studenta. Słupki błędów odzwierciedlają +/- SD (w celu interpretacji odniesień do koloru w legendzie tej figury czytelnik jest odsyłany do internetowej wersji tego artykułu).
Sulforaphane upośledza mitochondrialne stowarzyszenie Drp1
W oparciu o odkrycie, że leczenie SFN indukuje hiperfuzję mitochondrialną, stwierdziliśmy, że ten fenotyp był albo wynikiem nadmiernej aktywności fuzji, albo hamowania aktywności rozszczepienia. Aby rozróżnić te dwie możliwości, porównaliśmy morfologię peroksysomów w obecności i nieobecności SFN. Peroksysomy są podobne do mitochondriów, ponieważ są dynamicznymi organellami, których kształt i długość stale się zmieniają [44]. Peroksysomy zawierają zarówno Fis1 jak i Mff w ich zewnętrznej błonie, aw konsekwencji są celami dla rozszczepienia za pośrednictwem Drp1 [22], [23]. Jednak peroksysomy nie wykorzystują maszynerii fuzyjnej sieci mitochondrialnej iw konsekwencji nie ulegają fuzji [39]. Przeciwnie, rozszczepienie peroksysomalne przeciwstawia się wydłużaniu istniejących peroksyzomów przez de novo dodatek błon i białek [44]. Ponieważ w peroksysomach brakuje Mfn1 / 2 i OPA1, sądziliśmy, że jeśli SFN aktywuje maszynerię fuzyjną, zamiast hamować maszynerię rozszczepienia, długość peroksysomu nie zostanie naruszona. W komórkach traktowanych podłożem, peroksysomy są utrzymywane jako krótkie, okrągłe, punktowe organelle (Fig. 2, panele b i d). Jednak leczenie SFN zwiększało długość peroksysomu o ~ 2-krotnie w porównaniu do komórek kontrolnych (ryc. 2, panele f i h). Co więcej, wiele peroksyzomów zostało zaciśniętych w pobliżu środka, co wskazuje na potencjalną wadę pęknięcia (rys. 2, panel h, groty strzałek). Podobnie, peroksysomy w komórkach transfekowanych siRNA Drp1 były nienormalnie długie (Fig. 2, panele j i 1), potwierdzając, że Drp1 jest wymagany do rozszczepienia peroksysomalnego i sugeruje, że leczenie SFN powoduje fenotypy mitochondrialne i peroksysomalne przez zakłócanie mechanizmów rozszczepienia.
Figura 2 SFN indukuje wydłużenie peroksysomalne. (A) Komórki RPE-1 transfekowano 10 nM wskazanego siRNA i 3 dni później traktowano DMSO lub 50 µM SFN przez 4 godziny. Peroksysomy (zielony) znakowano przeciwciałem anty-PMP70, mitochondria MitoTracker (czerwony) i DNA barwiono kontrastowo DAPI. Powiększone wstawki peroksysomów pokazano po prawej stronie (panele d, h i l), aby ułatwić wizualizację zmian w morfologii wywołanych wyczerpaniem SFN i Drp1. Groty strzałek podkreślają punkty przewężenia. (W celu interpretacji odniesień do koloru w legendzie tej figury czytelnik jest odsyłany do internetowej wersji tego artykułu).
Następnie ustaliliśmy, w jaki sposób SFN ogranicza funkcję Drp1. Możliwości obejmowały zmniejszenie poziomu ekspresji, rekrutację/retencję w mitochondriach, oligomeryzację lub aktywność enzymatyczną GTPazy. Niedobór któregokolwiek z nich skutkowałby zmniejszeniem rozszczepienia mitochondriów i hiperfuzji. Nie wykryliśmy powtarzalnych zmian w poziomach białka Drp1 po leczeniu SFN (ryc. 1C i 3A), a zatem doszliśmy do wniosku, że SFN nie zmienia stabilności ani ekspresji Drp1, co jest zgodne z Drp1 mającym okres półtrwania >10 godzin [50] a nasze zabiegi SFN trwają krócej. Następnie zbadaliśmy, czy SFN wpływa na rekrutację lub retencję Drp1 w mitochondriach. Badania frakcjonowania wykazały, że SFN indukował utratę Drp1 z frakcji mitochondrialnej (ryc. 3A, ścieżki 7 i ryc. 8B). Jak informowaliśmy wcześniej [3], tylko niewielka część Drp43 (~1%) jest związana z siecią mitochondrialną w dowolnym momencie w warunkach stanu stacjonarnego, przy czym większość enzymu znajduje się w cytoplazmie (ryc. 3A, ścieżki 3 ). Te dane frakcjonowania zostały potwierdzone za pomocą analizy kolokalizacji, która wykazała ~5% redukcję w zlokalizowanych w mitochondriach, punktowych ogniskach Drp8 po traktowaniu SFN (ryc. 40C i D). Razem dane te wskazują, że fuzja mitochondrialna indukowana przez SFN jest, przynajmniej częściowo, spowodowana osłabionym powiązaniem Drp1 z mitochondriami. Nasze dane nie rozróżniają, czy SFN zakłóca rekrutację mitochondrialną czy mitochondrialną retencję Drp3, czy jedno i drugie, ponieważ analiza endogennego Drp1 nie była podatna na wizualizację GTPazy za pomocą mikroskopii żywych komórek.
Rysunek 3 SFN powoduje utratę Drp1 z mitochondriów. (A) Frakcjonowanie subkomórkowe komórek RPE-1 po 4 h DMSO lub SFN. Lizaty całych komórek (WCL), jądrowe (Nuc), cytozolowe (Cyto) i surowe frakcje mitochondrialne (Mito) rozdzielono za pomocą SDS-PAGE i poddano obróbce metodą Western blot wskazanymi przeciwciałami. Migracja markerów masy cząsteczkowej jest wskazana po lewej stronie. (B) Wykresy przedstawiające kwantyfikację densytometryczną Drp1 we wskazanych frakcjach z (A). (C) Komórki RPE-1 transfekowano 10 nM siCON lub siDrp1 i 3 dni później traktowano DMSO lub SFN dla 4 h. Drp1 (zielony) wizualizowano za pomocą przeciwciała anty-Drp1, mitochondriów z MitoTracker (czerwony) i jąder z DAPI (niebieski). (D) Zautomatyzowana analiza kolokalizacji sygnału Drp1 i MitoTracker z (C). Dane w (B) i (D) zebrano odpowiednio z niezależnych eksperymentów 3 i 5, a istotność statystyczną określono za pomocą dwustronnego testu t-Studenta. Słupki błędów odzwierciedlają +/- SD, a gwiazdki oznaczają istotność statystyczną. (W celu interpretacji odniesień do koloru w legendzie tej figury czytelnik odsyła do internetowej wersji tego artykułu).
Sulforaphane zapewnia ochronę przed apoptozą wywołaną przez staurosportinę Niezależnie od Nrf2
Wcześniejsze prace wykazały, że rozszczepienie mitochondriów umożliwia tworzenie porów w zewnętrznej błonie mitochondrialnej generowanych przez Bax/Bak podczas apoptozy [11]. Wykazano, że Drp1 jest selektywnie rekrutowany do mitochondriów podczas apoptozy [11] i, zgodnie z tym, fragmentaryczne mitochondria zaobserwowano na wczesnym etapie procesu [27]. I odwrotnie, uważa się, że hamowanie rozszczepienia mitochondriów hamuje apoptozę poprzez blokowanie tworzenia porów błony zewnętrznej, które umożliwiają uwalnianie cytochromu c [53]. W związku z tym stymulacja fuzji mitochondrialnej opóźnia postęp apoptozy indukowanej przez związki zawierające staurosporynę (STS) [14]. Aby określić, czy SFN chroni komórki RPE-1 przed apoptozą za pośrednictwem STS, a jeśli tak, czy wymaga to Nrf2, opracowaliśmy test do łatwej indukcji rozszczepiania polimerazy poli ADP rybozy (PARP), substrat aktywowanej kaspazy-3 i ostateczny marker apoptoza. Traktowanie komórek RPE-1 1 µM STS przez 6 godzin powodowało jedynie bardzo niewielkie rozszczepienie PARP, ale zapobiegało temu wspólne traktowanie SFN (np. Fig. 4A, ścieżka 3 w porównaniu z 4). Aby zwiększyć wiarygodność tego testu, dodatkowo uwrażliwiliśmy komórki na apoptozę indukowaną przez STS przez wstępne traktowanie ich siRNA ukierunkowanym na czynnik antyapoptotyczny, Bcl-XL. To wstępne traktowanie zmniejszyło ekspresję Bcl-XL i znacznie promowało rozszczepianie PARP w funkcji czasu ekspozycji na STS (ryc. 4B, porównaj ścieżkę 2 do ścieżek 4-10). Co ważne, 2 godziny wstępnego traktowania SFN złagodziły rozszczepienie PARP w komórkach wystawionych na STS (ryc. 4C, ścieżka 3 wobec 4 i ścieżka 5 wobec 6). Podobnie, komórki stabilnie zubożone w Nrf2 przez CRISPR/Cas9 były porównywalnie chronione przed toksycznością STS przez wstępne traktowanie SFN (Fig. 4C, ścieżka 11 wobec 12 i ścieżka 13 wobec 14 i Ryc. 4D). Ochronę tę obserwowano stosując jako odczyty zarówno rozszczepienie PARP (fig. 4C i D), jak i morfologię komórkową (fig. 4E). Skuteczność zubożenia Nrf2 przez CRISPR/Cas9 potwierdzono metodą western blot (ryc. 4C, nrf2 blot). Jak przewidywano, komórki zubożone w Drp1, które również dają fenotyp hiperfuzji (ryc. 1A), również blokowały cięcie PARP w odpowiedzi na STS w porównaniu z komórkami kontrolnymi inkubowanymi z SFN (ryc. 4F i G). Podsumowując, odkrycia te są zgodne z tym, że SFN nadaje ochronę przed apoptozą poprzez swoją zdolność do ograniczania funkcji Drp1, niezależnie od stabilizacji i aktywacji Nrf2.
Figura 4 Efekty cytoprotekcyjne SFN są niezależne od ekspresji Nrf2 (A) Komórki RPE-1 wstępnie traktowano DMSO lub 50 µM SFN przez 2 godziny przed traktowaniem DMSO, 1 µM staurosporyną (STS) lub 50 µM staurosporyną (STS) lub 6 µM SFN przez XNUMX godziny. M etopozyd przez XNUMX godzin i poddano obróbce do analizy metodą western blot anty-PARP. (B) Komórki RPE-1 transfekowano 2.5 nM siCON, 1 nM siBcl-XL lub 2.5 nM siBcl-XL i 3 dni później traktowano DMSO lub 1 M STS przez 2, 4 lub 6 godzin. Przedstawiono reprezentatywne western bloty, a migrację markerów masy cząsteczkowej wskazano po lewej stronie. (C) Komórki RPE-9 generowane przez CRISPR / Cas2 typu dzikiego (Nrf2WT) i Nrf2 knockout (Nrf1KO) transfekowano 1 nM siBcl-XL i 3 dni później wstępnie traktowano DMSO lub 50 μM SFN przez 2 godziny . Następnie komórki traktowano 1 µM STS przez 2, 4 lub 6 godzin. Przedstawiono reprezentatywne analizy western blot ze wskazanymi przeciwciałami. (D) Kwantyfikacja odszczepionego PARP jako procent całkowitej PARP (odszczepiony + nierozszczepiony) z 3 niezależnych eksperymentów. Co ważne, poziomy rozszczepionego PARP były porównywalne niezależnie od tego, czy komórki wyrażały Nrf2, czy nie, co wskazuje, że ochrona SFN przed STS jest niezależna od czynnika transkrypcyjnego. (E) Obrazy z kontrastem fazowym 20X wykonane bezpośrednio przed zebraniem lizatów z (C). Skala = 65 m. (F) Reprezentatywne western blot wykazujące, że zubożenie Drp1 zapewnia prawie porównywalną ochronę przed STS jak traktowanie SFN. Komórki RPE-1 transfekowano 1 nM siBcl-XL i dodatkowo transfekowano 10 nM siCON lub 10 nM siDrp1. 3 dni później komórki siCON traktowano wstępnie SFN jak w (A) i (C), a następnie eksponowano na STS przez 4 godziny przed zebraniem i obróbką do analizy western blot ze wskazanymi przeciwciałami. (G) To samo co (D) dla danych przedstawionych w (F) zebranych z 3 niezależnych eksperymentów. Słupki błędów odzwierciedlają +/- SEM
Dyskusja
Odkryliśmy, że SFN moduluje mitochondrialne rozszczepienie / dynamikę fuzji niezależnie od jej wpływu na szlak KEAP1-Nrf2-ARE. Jest to intrygujące z powodu założonego związku między dysfunkcją mitochondriów i produkcją ROS oraz koniecznością zrekompensowania wolnych rodników mitochondriów poprzez aktywację Nrf2. Ten dodatkowy, funkcjonalny wpływ SFN ma potencjalne znaczenie, biorąc pod uwagę, że obecnie trwają obecnie badania kliniczne 30, które testują SFN w leczeniu różnych chorób, w tym raka gruczołu krokowego, obturacyjnej choroby płuc i choroby sierpowatokrwinkowej [7], [10], [ 47].
Ponieważ SFN jest izotiocyjanianem [56] i aktywuje sygnalizację Nrf2 poprzez bezpośrednie acylowanie krytycznych cystein KEAP1 w celu zahamowania degradacji Nrf2 [21], wynika z tego, że SFN wywiera działanie profuzyjne poprzez modulowanie aktywności czynnika rozszczepienia lub fuzji poprzez modyfikację cysteiny . Nasze dane silnie potwierdzają, że Drp1 jest ujemnie regulowany przez SFN, chociaż to, czy GTPaza jest bezpośrednim celem acylacji, pozostaje do wyjaśnienia. Pomimo tej luki w wiedzy, funkcja Drp1 jest wyraźnie zagrożona przez SFN, ponieważ zarówno mitochondria, jak i peroksysomy ulegają nadmiernej fuzji w odpowiedzi na leczenie SFN, a te organelle dzielą Drp1 dla odpowiednich zdarzeń rozszczepienia [38]. Ponadto SFN zmniejsza ilość Drp1, która lokalizuje i gromadzi się w mitochondriach (ryc. 3). Ponieważ nasze eksperymenty przeprowadzono ze wszystkimi białkami endogennymi, nasze wykrywanie Drp1 w mitochondrialnych miejscach rozszczepienia odbywa się w warunkach stanu stacjonarnego, a w konsekwencji nie możemy odróżnić defektu rekrutacji od retencji enzymu spowodowanego przez SFN. Co więcej, nie możemy jeszcze wyeliminować możliwości, że SFN acyluje receptor w mitochondriach (Fis1 lub Mff), aby zablokować rekrutację Drp1, podejrzewamy, że Drp1 jest bezpośrednio modyfikowany. Drp1 ma dziewięć cystein, z których osiem znajduje się w domenie środkowej, która jest wymagana do oligomeryzacji [3], a jedna z nich znajduje się w domenie efektorowej GTPazy (GED) na C-końcu Drp1. Bezpośrednia acylacja którejkolwiek z tych cystein może spowodować defekt aktywności w Drp1, a zatem leży u podstaw wpływu SFN na dynamikę mitochondriów. Warto zauważyć, że wcześniejsze prace sugerują, że defekty w oligomeryzacji i aktywności katalitycznej mogą znosić retencję Drp1 w mitochondriach [52]. Cys644 w domenie GED jest szczególnie atrakcyjnym celem na podstawie wcześniejszych prac wykazujących, że mutacja tej fenokopii cysteiny zaburza aktywność GTPazy Drp1 [4] i że ta konkretna cysteina jest modyfikowana przez elektrofile reagujące z tiolami [9]. Rozwiązanie tej nierozstrzygniętej kwestii będzie wymagało walidacji metodą spektrometrii mas. Podsumowując, zidentyfikowaliśmy nową, cytoochronną funkcję istotnego klinicznie związku SFN. Oprócz aktywacji głównego antyoksydacyjnego czynnika transkrypcyjnego Nrf2, SFN promuje fuzję mitochondrialną i peroksysomalną, a efekt ten jest niezależny od Nrf2. Mechanizm leżący u podstaw tego zjawiska polega na zmniejszeniu funkcji GTPazy Drp1, głównego mediatora rozszczepienia mitochondriów i peroksysomów. Główną konsekwencją fuzji mitochondrialnej za pośrednictwem SFN jest to, że komórki stają się oporne na toksyczne działanie staurosporyny, induktora apoptozy. To dodatkowe cytoochronne działanie SFN może mieć szczególne zastosowanie kliniczne w licznych chorobach neurodegeneracyjnych, w przypadku których wiek jest głównym czynnikiem ryzyka (np. choroba Parkinsona, choroba Alzheimera, zwyrodnienie plamki żółtej związane z wiekiem), ponieważ choroby te są związane z apoptozą i zmniejszoną poziom i/lub rozregulowanie Nrf2 [35], [36], [48].
Materiały i Metody
Testy apoptozy
Komórki wysiano i transfekowano siRNA, jak wskazano poniżej. Komórki wstępnie traktowano 50 µM sulforafanem przez 2 godziny w celu wywołania fuzji mitochondrialnej, a następnie traktowano 1 µM staurosporyną w celu wywołania apoptozy. W czasie zbioru pożywki zbierano do pojedynczych probówek i poddawano wirowaniu z dużą prędkością w celu osadzenia komórek apoptotycznych. Ten osad komórek połączono z adherentnymi komórkami i rozpuszczono w 2-krotnie stężonym buforze Laemmli. Próbki poddano western blottingowi anty-PARP.
CRISPR / Cas9 Construct Generation
Aby utworzyć LentiCRISPR / eCas9 1.1, LentiCRISPR v2 (addgene #52961) został po raz pierwszy wycięty za pomocą Age1 i BamH1. Następnie, SpCas9 z eSpCas9 1.1 (addgene # 71814) poddano amplifikacji PCR z nawisami Age1 i BamH1 przy użyciu następujących starterów (Forward AGCGCACCGGTTCTAGAGCCTCTACCCACCATGGACTATAAGGACCACGAC, Reverse AAGCGCGGATCCCTTTTTCTTTTTTGCCTGGCCGG) i zligowano do wektora cięcia powyżej. Sekwencje sgRNA określono za pomocą Benchling.com. Parametry zostały ustawione tak, aby były ukierunkowane na sekwencję kodującą o najwyższej wartości docelowej i najniższej wartości docelowej. Następujące sekwencje (sekwencję kierującą podkreślone HS sgNFE2L2 # 1 sens CACCGCGACGGAAAGAGTATGAGC antysensownej AAACGCTCATACTCTTTCCGTCGC; HS sgNFE2L2 # 2 poczucie CACCGGTTTCTGACTGGATGTGCT antysensownej AAACAGCACATCCAGTCAGAAACC; hs sgNFE2L2 # 3 sens CACCGGAGTAGTTGGCAGATCCAC antysensownej AAACGTGGATCTGCCAACTACTCC) hybrydyzowano i poddano ligacji z BsmB1 cięcia LentiCRISPR / eCas9 1.1. Komórki RPE-1 zakażone lentiwirusowo wybrano puromycyną i utrzymywano jako populację zbiorczą. Knockout został potwierdzony przez immunofluorescencję i western blotting.
Hodowla komórek i transfekcje
Ludzkie komórki nabłonka barwnikowego siatkówki transformowane telomerazą (RPE-1) (ATCC) hodowano w pożywce Dulbecco's Modified Eagle Medium (DMEM) zawierającej 1 g / l glukozy z dodatkiem penicyliny, streptomycyny, 1X nieistotnego koktajlu aminokwasowego (Life Technologies), i 10% płodowej surowicy bydlęcej (Life Technologies). W przypadku transfekcji siRNA przez noc wysiano 30,000 komórek / ml. Komórki otrzymały 35,000 nM siRNA rozcieńczonego w wolnym od surowicy DMEM i połączone z 10% odczynnikiem do transfekcji interferiny (PolyPlus). W celu uczulenia na apoptozę komórki otrzymały 0.3 nM siRNA Bcl-XL. Komórki zebrano 1-2 dni po transfekcji.
Chemikalia, przeciwciała i siRNA Oligos
Przeciwciała przeciwko? -Tubulinie (Cell Signaling),? -Tubulinie (Sigma), Drp1 (BD Biosciences), KEAP1 (Proteintech), Lamin B1 (Abcam), PARP (Cell Signaling), PMP70 (Abcam) i Tom20 (BD Biosciences) ) zastosowano w rozcieńczeniach 1: 1000 do analizy western blot i immunofluorescencji. Własne, królicze przeciwciało anty-Nrf2 zastosowano w stosunku 1: 2000 do western blotting [34], [59]. Zastosowano sulforafan (Sigma) i staurosporynę (Tocris) w stężeniu odpowiednio 50 µM i 1 µM. siRNA przeciwko Drp1 (Dharmacon), Nrf2 (Dharmacon), KEAP1 (Cell Signaling) i Bcl-XL (Cell Signaling) zastosowano przy 10 nM, chyba że zaznaczono inaczej.
Immunofluorescencja i znakowanie Vivo
Komórki wysiane na 18 mm szkiełkach nakrywkowych traktowano nośnikiem lub lekiem, utrwalano w 3.7% formaldehydzie, a następnie permeabilizowano w 0.2% Triton X-100 / PBS na lodzie przez 10 minut. Pierwotne przeciwciała inkubowano w 3% albuminie surowicy bydlęcej (BSA) w PBS przez noc w 4 ° C. Po przemyciu PBS komórki inkubowano przez 1 godzinę w odpowiednich dla gatunku, sprzężonych przeciwciałach drugorzędowych Alexa488 lub Alexa546 (rozcieńczone 1: 1000) i 0.1 μg / ml DAPI (Sigma) w 3% BSA / PBS. Mitochondria wizualizowano albo za pomocą immunofluorescencji anty-Tom20, albo przez inkubację komórek w 200 nM MitoTracker Red CMXRos (Molecular Probes, Inc.) w wolnym od surowicy DMEM przez 30 minut w 37 ° C przed utrwaleniem.
Mikroskopia i analiza obrazu
Próbki immunofluorescencyjne oglądano na mikroskopie konfokalnym LSM710 (Carl Zeiss). Mikrofotografie wykonano przy użyciu obiektywów do immersji w olejku 63X lub 100X, a obrazy dostosowano i ulepszono za pomocą programu Adobe Photoshop CS6. Analiza kolokalizacji została przeprowadzona przy użyciu funkcji kolokalizacji Carl Zeiss LSM710 z ręcznie ustawionymi progami bez znajomości tożsamości próbek. Słupki skali na całej długości, o ile nie wskazano inaczej, wynoszą 10 µm. Morfologię mitochondriów oceniano metodą ślepej punktacji. Jeśli mitochondria komórki były utrzymywane jako wielokrotne, okrągłe, rozróżniające puncta, komórka była oceniana jako `` rozszczepienie ''. Jeśli poszczególne mitochondria były nierozróżnialne, a cała sieć mitochondrialna wydawała się ciągła, komórka była oceniana jako `` fuzja ''. Wszystkie inne komórki, w tym te ze skupiającymi się mitochondriami, zostały ocenione jako `` pośrednie ''.
Frakcje subkomórkowe
Komórki RPE-1 hodowano do konfluencji. Po przemyciu PBS komórki poddawano wirowaniu przy 600 µg przez 10 minut i ponownie zawieszano w 600 µl buforu do izolacji (210 mM Mannitol, 70 mM Sacharoza, 5 mM MOPS, 1 mM EDTA pH 7.4 + 1 mM PMSF). Zawiesinę poddano 30-krotnej lizie w homogenizatorze Dounce. Część homogenatu została zakonserwowana jako `` lizat całych komórek ''. Pozostałość poddano wirowaniu przy 800 g przez 10 minut w celu zarodkowania osadu. Supernatanty poddano wirowaniu przy 1500 g przez 10 minut w celu oczyszczenia pozostałych jąder i niezlizowanych komórek. Ten supernatant poddano wirowaniu przy 15,000 15 g przez XNUMX minut w celu osadzenia mitochondriów. Supernatant został zachowany jako `` frakcja cytozolowa ''. Osad delikatnie przemyto PBS i ponownie zawieszono w buforze do izolacji. Stężenie białka w każdej frakcji mierzono za pomocą testu z kwasem bicynchoninowym (BCA) i równoważne ilości białka rozdzielano za pomocą SDS-PAGE.
Western Blotting
Komórki przepłukano w PBS i rozpuszczono w 2-krotnie stężonym buforze solubilizującym Laemmli (100 mM Tris [pH 6.8], 2% SDS, 0.008% błękitu bromofenolowego, 2% 2-merkaptoetanolu, 26.3% glicerolu i 0.001% piryniny Y). Lizaty gotowano przez 5 minut przed nałożeniem na żele poliakryloamidowe z dodecylosiarczanem sodu (SDS). Białka przeniesiono na membrany nitrocelulozowe i membrany zablokowano przez 1 godzinę w 5% mleku/TBST. Pierwotne przeciwciała rozcieńczono w 5% mleku/TBST i inkubowano z odciskiem przez noc w temperaturze 4°C. Przeciwciała drugorzędowe skoniugowane z peroksydazą chrzanową (HRP) rozcieńczono w 5% mleku/TBST. Bloty przetwarzano z ulepszoną chemiluminescencją, a kwantyfikację densytometryczną przeprowadzono przy użyciu oprogramowania ImageJ.
Sulforafan jest substancją chemiczną pochodzącą z kolekcji izotiocyjanianów substancji organosiarczkowych otrzymywanych między innymi z warzyw krzyżowych, w tym brokułów, kapusty, kalafiora, jarmużu i kapusty. Sulforafan jest wytwarzany, gdy enzym myrosinase przekształca glukorafaninę, glukozynolan, w sulforafan, znany również jako sulforafanoglukozynolan. Kiełki brokułów i kalafior mają najwyższe stężenie glukorafaniny lub prekursora sulforafanu. Badania wykazały, że sulforafan zwiększa zdolności antyoksydacyjne organizmu ludzkiego, aby zapobiegać różnym problemom zdrowotnym. Dr Alex Jimenez DC, CCST Insight
Sulforafan i jego wpływ na raka, śmiertelność, starzenie się, mózg i zachowanie, choroby serca i nie tylko
Izotiocyjaniany to jedne z najważniejszych związków roślinnych, które możesz znaleźć w swojej diecie. W tym filmie przedstawiam dla nich najbardziej wyczerpujący argument, jaki kiedykolwiek powstał. Krótki czas koncentracji? Przejdź do swojego ulubionego tematu, klikając jeden z poniższych punktów czasowych. Pełna oś czasu poniżej.
Kluczowe sekcje:
00: 01: 14 - Rak i śmiertelność
00: 19: 04 - Starzenie
00: 26: 30 - Mózg i zachowanie
00: 38: 06 - Ostatnie podsumowanie
00: 40: 27 - Dawka
Pełna oś czasu:
00: 00: 34 - Wprowadzenie sulforaphane, główny punkt widzenia wideo.
00: 01: 14 - spożycie warzyw krzyżowych i redukcja śmiertelności z wszystkich przyczyn.
00: 02: 12 - ryzyko raka prostaty.
00: 02: 23 - ryzyko raka pęcherza moczowego.
00: 02: 34 - Rak płuc w ryzyku palaczy.
00: 02: 48 - ryzyko raka piersi.
00: 03: 13 - Hipotetyczny: co, jeśli masz już raka? (interwencyjne)
00:03:35 – Prawdopodobny mechanizm napędzający dane asocjacyjne dotyczące raka i śmiertelności.
00: 04: 38 - Sulforafan i rak.
00:05:32 – Dowody na zwierzętach wykazujące silny wpływ ekstraktu z kiełków brokułów na rozwój guza pęcherza moczowego u szczurów.
00: 06: 06 - Wpływ bezpośredniej suplementacji sulforafanu u pacjentów z rakiem prostaty.
00: 11: 20 - napój z kiełków brokułów zwiększa wydalanie benzenu o 61%.
00: 13: 31 - homogenat z kiełków brokułów zwiększa enzymy antyoksydacyjne w górnych drogach oddechowych.
00: 15: 45 - śmiertelność wśród roślin krzyżowych i śmiertelność z powodu chorób serca.
00: 16: 55 - proszek z kiełków brokułów poprawia poziom lipidów we krwi i ogólne ryzyko chorób serca u chorych na cukrzycę typu 2.
00:19:04 – Początek sekcji starzenia.
00:19:21 – Dieta wzbogacona w sulforafan wydłuża życie chrząszczy od 15 do 30% (w określonych warunkach).
00: 20: 34 - Znaczenie niskiego stanu zapalnego dla długowieczności.
00: 22: 05 - warzywa kapustne i kiełki w proszku z brokułów wydają się zmniejszać szeroki zakres markerów stanu zapalnego u ludzi.
00: 23: 40 - Podsumowanie filmu wideo: rak, sekcje starzenia
00: 24: 14 - Badania na myszach sugerują, że sulforafan może poprawić zdolność adaptacyjną układu odpornościowego w starszym wieku.
00:25:18 – Sulforafan poprawił wzrost włosów w mysim modelu łysienia. Zdjęcie o 00:26:10.
00: 26: 30 - Początek sekcji mózgu i zachowania.
00: 27: 18 - Wpływ wyciągu z kiełków brokułów na autyzm.
00: 27: 48 - Wpływ glukorafaniny na schizofrenię.
00: 28: 17 - Rozpoczęcie dyskusji o depresji (prawdopodobny mechanizm i badania).
00:31:21 – Badanie na myszach przy użyciu 10 różnych modeli depresji wywołanej stresem wykazało, że sulforafan jest podobnie skuteczny jak fluoksetyna (prozac).
00: 32: 00 - Badanie pokazuje, że bezpośrednie spożycie glukorafaniny u myszy jest podobnie skuteczne w zapobieganiu depresji w modelu stresu społecznego.
00: 33: 01 - Początek sekcji zwanej neurodegeneracją.
00: 35: 01 - Sulforafan wstrzyknięty natychmiast po TBI poprawia pamięć (badanie myszy).
00: 35: 55 - Sulforafan i plastyczność neuronalna.
00:36:32 – Sulforafan poprawia uczenie się w modelu cukrzycy typu II u myszy.
00:37:19 – Dystrofia mięśniowa Sulforaphane i Duchenne'a.
00: 37: 44 - Hamowanie miostatyny w komórkach satelitarnych mięśni (in vitro).
00: 38: 06 - późne nagranie wideo: śmiertelność i rak, uszkodzenie DNA, stres oksydacyjny i zapalenie, wydalanie benzenu, choroba sercowo-naczyniowa, cukrzyca typu II, wpływ na mózg (depresja, autyzm, schizofrenia, neurodegeneracja), szlak NRF2.
00: 40: 27 - Zastanawiasz się nad określeniem dawki kiełków brokułów lub sulforafanu.
00: 41: 01 - Anegdoty dotyczące kiełkowania w domu.
00: 43: 14 - Przy temperaturach gotowania i aktywności sulforafanowej.
00: 43: 45 - Transformacja bakterii jelitowych sulforafanu z glukorafaniny.
00: 44: 24 - Suplementy działają lepiej w połączeniu z aktywną miozynazą z warzyw.
00: 44: 56 - Techniki gotowania i warzywa kapustne.
Ogrzewanie Zmniejsza aktywność białka epithiospecyfikatora i zwiększa tworzenie sulforafanu w brokułach
Abstrakcyjny
Sulforafan, izotiocyjanian brokułów, jest jednym z najsilniejszych środków przeciwrakowych pochodzących z żywności. Związek ten nie występuje w nienaruszonym warzywie, lecz powstaje z jego prekursora glukozynolanów, glukorafaniny, w wyniku działania mirozynazy, enzymu tioglukozydazy, podczas miażdżenia lub żucia tkanki brokułów. Jednak wiele badań wykazało, że wydajność sulforafanu z glukorafaniny jest niska, a niebioaktywny analog nitrylu, nitryl sulforafanu, jest głównym produktem hydrolizy, gdy tkanka roślinna jest rozdrabniana w temperaturze pokojowej. Ostatnie dowody sugerują, że u Arabidopsis tworzenie nitrylu z glukozynolanów jest kontrolowane przez białko wrażliwe na ciepło, białko epitiospecyfikatora (ESP), niekatalityczny kofaktor mirozynazy. Naszym celem było zbadanie wpływu ogrzewania różyczek i kiełków brokułów na tworzenie nitrylu sulforafanu i sulforafanu, ustalenie, czy brokuły zawierają aktywność ESP, a następnie skorelowanie zależnych od ciepła zmian w aktywności ESP, zawartości sulforafanu i bioaktywności, mierzonych przez indukcję enzym detoksykacyjny fazy II, reduktaza chinonowa (QR) w hodowli komórkowej. Ogrzewanie świeżych różyczek brokułów lub kiełków brokułów do 60 °C przed homogenizacją jednocześnie zwiększyło tworzenie sulforafanu i zmniejszyło tworzenie sulforafanu i nitrylu. Znacząca utrata aktywności ESP zbiegła się ze spadkiem tworzenia nitrylu sulforafanowego. Ogrzewanie do 70°C i powyżej zmniejszyło powstawanie obu produktów w różyczkach brokułów, ale nie w kiełkach brokułów. Indukcja QR w hodowanych mysich komórkach wątrobiaka Hepa lclc7 równolegle zwiększa tworzenie sulforafanu.
Wstępne podgrzanie różyczek i kiełków brokuła do 60 ° C znacznie zwiększyło katalizowane przez mirozynazę tworzenie sulforafanu (SF) w ekstraktach z tkanek roślinnych po zmiażdżeniu. Było to związane ze spadkiem tworzenia sulforafanonitrylu (SF Nitryl) i aktywności białka epithiospecifier (ESP).
Podsumowując, sulforafan to fitochemikalia występująca w brokułach i innych warzywach z rodziny krzyżowych. Niekontrolowana ilość utleniaczy spowodowana zarówno czynnikami wewnętrznymi, jak i zewnętrznymi może powodować stres oksydacyjny w ludzkim ciele, co może ostatecznie prowadzić do różnych problemów zdrowotnych. Sulforafan może aktywować produkcję Nrf2, czynnika transkrypcyjnego, który pomaga regulować ochronne mechanizmy antyoksydacyjne, które kontrolują odpowiedź komórki na utleniacze. Zakres naszych informacji ogranicza się do kwestii chiropraktyki i zdrowia kręgosłupa. Aby omówić ten temat, prosimy zapytać dr Jimeneza lub skontaktować się z nami pod adresem�915-850-0900 .
Na ból plecówTo jedna z najczęstszych przyczyn niepełnosprawności i opuszczonych dni w pracy na całym świecie. Ból pleców to druga najczęstsza przyczyna wizyt lekarskich, przewyższająca liczebnie jedynie infekcje górnych dróg oddechowych. Około 80 procent populacji przynajmniej raz doświadczy bólu pleców w ciągu swojego życia. Kręgosłup to złożona struktura składająca się z kości, stawów, więzadeł i mięśni oraz innych tkanek miękkich. Z tego powodu urazy i / lub zaostrzone warunki, takie jak przepukliny, może ostatecznie prowadzić do objawów bólu pleców. Urazy sportowe lub wypadki samochodowe są często najczęstszą przyczyną bólu pleców, jednak czasami najprostsze ruchy mogą mieć bolesne skutki. Na szczęście alternatywne opcje leczenia, takie jak opieka chiropraktyczna, mogą pomóc złagodzić ból pleców poprzez zastosowanie korekcji kręgosłupa i manualnych manipulacji, ostatecznie poprawiając ulgę w bólu.
Narzędzie Find A Practitioner firmy IFM to największa sieć skierowań w medycynie funkcjonalnej, stworzona, aby pomóc pacjentom zlokalizować lekarzy medycyny funkcjonalnej na całym świecie. Certyfikowani praktycy IFM są wymienieni na pierwszym miejscu w wynikach wyszukiwania ze względu na ich rozległe wykształcenie w zakresie medycyny funkcjonalnej